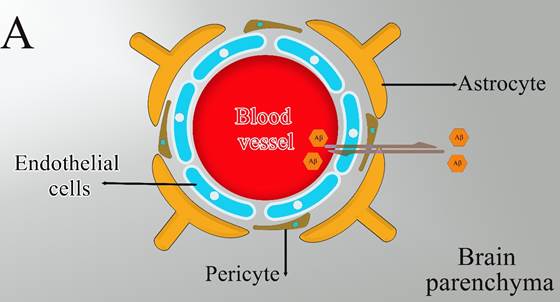
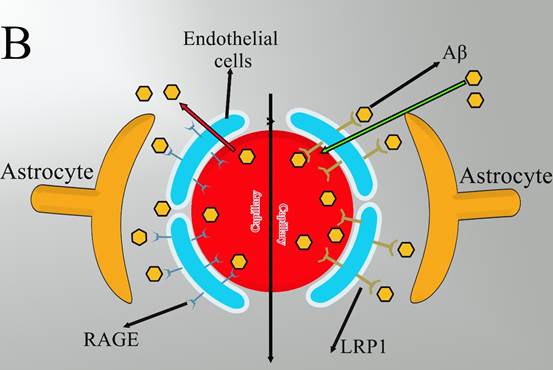
Figure 1. The proposed mechanism where
astrocytes are associated with Aβ clearance).
The role of
astrocytes in Alzheimer's disease, A systematic review
Seyyed
Mohammad Taghi Razavi-Toosi 1,2, Parvin Babaei 3,4,
Arefeh Salehi 5*
1 Medical Biotechnology Research Center, School of Paramedicine, Guilan
University of Medical Sciences, Rasht, Iran
2 Cardiovascular Diseases Research Center, Department of Cardiology,
Heshmat Hospital, School of Medicine, Guilan University of Medical Sciences,
Rasht, Iran
3 Neuroscience Research Center, Guilan University of Medical Sciences,
Rasht, Iran
4 Cellular & Molecular Research Center, Guilan University of Medical
Sciences, Rasht, Iran
5 Neurophysiology Research Centre, School of Medicine, Shahid Beheshti
University of Medical Sciences, Tehran, Iran
*Corresponding
Author: Arefeh
Salehi
* Email: arefehsalehi92@gmail.com
Abstract
Introduction: Alzheimer's disease (AD), the most common neurodegenerative disease in
the world, appears in two forms, early and late. Pathologically, an amyloid
beta peptide is the hallmark of this disease which is followed by synaptic
dysfunction, brain atrophy, and accumulation of neuronal tangles. The purpose
of this study is to review the researchers on astrocytes' role in the progress
of AD.
Materials
and Methods: A comprehensive search was conducted in databases articles focusing on
key terms "Inflammatory reactions", "Alzheimer's disease",
"Inflammatory factors" and "Astrocytes" and Boolean
operators. Articles before 2001 were removed.
Results: Finally, after analyzing the selected articles, 20 articles were
extracted and included in this review.
Conclusion: Astrocytes are a group of glial cells in the central nervous system. The
inflammatory activity of astrocytes plays a role in the development and
progression of Alzheimer's disease. They
strengthen the function of synapses by secreting neurotrophic factors. They also clear amyloid beta peptides from
nerve tissue. Amyloid beta peptides bind to specific
receptors on these cells and change the activity of these cells from
anti-inflammatory to inflammatory type. It seems that astrocytes play a
pivotal role in the development and progression of AD, particularly at the late
stage of the disease. Finding a rational strategy to suppress inflammatory A1
phenotype might be a promising tool to slow down the progress of AD.
Keywords: Alzheimer's disease, Astrocytes, Inflammatory factors, Amyloid beta
Introduction
Alzheimer's
disease (AD) is the most important and common neurodegenerative disease in the
world. Global statistics state that in 2017,
about 44 million people were affected by this disease. In the United States, AD is the only disease without a cure
among the 10 leading causes of human death.
In 2017, the costs paid in America for these patients were 259 billion dollars.
It is predicted that by 2050, these costs can increase to an impressive figure
of 1.1 trillion dollars (1, 2). This disease
exists in two forms: early or familial and late sporadic (3). The late type affects individuals
over 65 years old, and the early type includes a small number of affected
people and occurs under 65 years of age (4).
Currently, the amyloid beta hypothesis stands as the most accepted hypothesis
which states that amyloid beta (Aβ)
peptides are the early finding in the brain of affected people. Therefore,
excessive accumulation of amyloid peptides in the form of amyloid plaques in
the brain tissue disturbed neural connections and initiates neuro inflammation
however, in normal brain Aβ is destroyed by various factors
such as neprilysin, endothelin-converting enzyme, insulin-degrading enzyme,
angiotensin-converting enzyme, plasmin and cathepsin D (5-8).
Other
important symptoms of this disease include functional disorders of synapses,
brain atrophy, and the creation of neuronal filament coils inside nerve cells,
which consist of tau-hyperphosphorylated protein (1, 2).
Despite all the efforts made in the field of
understanding this disease and the factors responsible for initiating AD, a
suitable and guaranteed treatment has not yet been provided. Therefore finding a new strategy to control the disease and
prevent its progression has great importance (9, 10).
Materials and Methods
A
complete and comprehensive search was conducted in the literature available in
PubMed, Scopus, and Google Scholar databases, and articles were searched using
the key terms "inflammatory reactions", "Alzheimer's
disease", "inflammatory factors" and "astrocytes".
Key
terms were selected using MeSH and Boolean operators such as "AND",
"OR" and "NOT" were used to connect these terms. From
October 2021 to December 2022, two researchers searched independently.
Results
In
this study, articles on Alzheimer's, inflammatory cytokines, memory, and
astrocytes were selected. In the following, the articles that were presented
about inflammatory diseases, brain, and depression, and also the articles
before 2001 were removed. Also, to avoid excluding other valuable studies, a
search was conducted to extract other related studies Abstract. Finally, 20
studies were extracted and included in this review.
Discussion
Astrocytes
are a group of glial cells present in the central nervous system (CNS) (11). These cells play important and
different roles in the CNS. Perhaps their most important role is to initiate
immune and inflammatory responses to prevent possible damage to nerve tissue.
Astrocytes are the main regulators of magnesium concentration in the brain (11). Along with pre-synaptic and
post-synaptic neurons, they are the main components of synapses and play a role
in regulating synaptic plasticity by secreting gliotransmitter (12, 13).
Astrocytic
dysfunction results in the failure of Aβ clearance.
The
balance between the production and clearance of Aβ plays a detrimental role in
AD, and an inefficient Aβ clearance may be more susceptible to AD (14). An increasing number of studies
have evidenced that astrocytes act as a cellular player in Aβ clearance and
degradation from the brain parenchyma into the perivascular space, across BBB
(Figure 1), or by enzymatic degradation (15).
The
BBB would be a diffusion barrier that impedes the influx into the brain
parenchyma of certain molecules based on polarity and size. The principal
cellular constituents of the BBB include capillary endothelial cells,
perivascular pericytes, and astrocyte end-feet (Figure
1A). Maintaining the normal physiological function of astrocytes will have a
critical role in the transport of Aβ across BBB into the circulation which is
mainly mediated by receptor for advanced glycation end products (RAGE) and
lipoprotein receptor-related protein 1 (LRP1) in endothelial cells (16). Since RAGE acts as an important
transporter via regulating the influx of circulating Aβ into the brain while
the efflux of brain-derived Aβ into the circulation via BBB is implemented by
LRP1 (14) (Figure 1B). In addition to the
direct factor that astrocytic dysfunction leads to the failure transport of Aβ
across BBB, astrocytic dysfunction may indirectly result in other avenues which
are associated with the failure of Aβ clearance from the brain, such as
abnormal interstitial fluid drainage and the failure of microglial phagocytosis
(17). Astrocytic dysfunction probably
induces the occurrence of neuroinflammation and oxidative stress, and then both
neuroinflammation and oxidative stress contribute to abnormal interstitial
fluid drainage and the failure of microglial phagocytosis, and the failure of
Aβ clearance, finally (18).
Figure 1. The proposed mechanism where
astrocytes are associated with Aβ clearance).
Major
Roles of Astrocytes in Alzheimer´s Disease
Alzheimer’s
disease (AD) is characterized by amyloid beta accumulation (Aβ or senile
plaques), formation of hyperphosphorylated tau neurofibrillary tangles,
neuroinflammation, synaptic demise, neuronal death, and brain dysfunction
leading to severe cognitive impairment. The amyloid hypothesis originally
postulated a linearity of progression according to Aβ accumulation, which
subsequently led to the formation of tangles and other pathological hallmarks (19). The role of glial cells, and
astrocytes in particular, in the neuropathology of many neurodegenerative
diseases, is universally acknowledged (20).
The
risk of AD is associated with genes mainly expressed by glial cells, either
astrocytes, microglia, and/or oligodendrocytes (21).
Apolipoprotein
E (APOE), a major genetic risk factor in Late-Onset AD (LOAD), is mainly
expressed in astrocytes in the healthy brain (22) and contributes to the accumulation
of Aβ in the brain (23).
Other
genes associated with AD such as Clusterin (CLU) and Fermitin family member 2
(FERMT2) are similarly predominantly expressed by astrocytes. Reactive
astrogliosis is prominent in AD being an early event in human patients and in
animal models, possibly even preceding the formation of Aβ Aβ
plaques
(24).
These
data suggest a crucial role of astrocytes in the pathogenesis of AD.
Morphological studies in post-mortem AD patient brains demonstrated close
interaction between astrocytes and Aβ depositions (25).
It is however unclear
how this close interaction translates into the disease progression.
Astrocytes, when associated with senile plaques, become reactive with
morphological hypertrophy manifested by thicker processes and increased
expression of the intermediate filament proteins glial fibrillary acidic
protein (GFAP), vimentin, nestin, and synemin (26).
Reactive
astrocytes are found in both human AD patient brains [75] and AD mice models (27)
Pathological
signals inducing astrogliosis in AD can be associated with damaged cells; Aβ by
itself is a strong instigator of astrocyte reactivity. At the molecular level,
Aβ induction of astrogliosis remodeling is mediated by Ca2+ release
from the endoplasmic reticulum; inhibition of the latter suppresses astrocytic
reactivity (28).
In
AD, astrocytes undergo relatively mild isomorphic gliosis and astrocytic
domains do not overlap, potentially indicating a defensive nature of the
astrocytic response. Indeed, inhibition of astrogliosis exacerbates Aβ accumulation
and histopathology in AD mice (29). Reactive astrocytes in the
vicinity of plaques display aberrant calcium dynamics (30).
In particular, human AD brains are
characterized by severe disruption or even complete disappearance of
interlaminar astrocytes (31). Atrophic astrocytes are
characterized by reduced volume and thinner processes. In the 3xTg-AD mice
model, atrophic astrocytes appear as early as 1 month of age in the entorhinal
cortex (EC), and the atrophy is sustained after 12 months of age when Aβ plaques
begin to appear (32).
Human
astrocytes derived from induced pluripotent stem cells (iPSC) from patients
with both familial and sporadic forms of AD also show atrophic phenotypes in
vitro compared to control cells (33).
While
atrophy might lead to loss of astrocyte homeostatic functions and give rise to
synaptic dysfunction, increased excitability, and/or damage of the BBB, (Figure
2) very little functional data are available. Finally, the neurodegenerative
process may directly damage astrocytes resulting
in
clasmatodendrosis, characterized by fragmentation and disappearance of distal
fine processes, along with swelling and vacuolation of the cell body (34) (Figure 2).
Astrocytes
could be, in principle, involved in Aβ production as they upregulate
β-secretase 1 and the amyloid precursor protein (APP) in the diseased brain (35).
However
no quantitative data points to astrocytes as the major source of Aβ. Astrocytes
are more likely to participate in Aβ clearance and elimination by different
mechanisms. Astrocytes express aquaporin 4 (AQP4) water channels in their
vascular end-feet and play an essential role in the glymphatic system
implicated in the clearance of Aβ (36) (Figure 2).
They
also produce amyloid beta-degrading proteases that cleave the peptide into
smaller fragments. The metalloendopeptidases neprilysin (NEP),
insulin-degrading enzyme (IDE), and endothelin-converting enzymes 1 and 2 (ECE1
and ECE2) are all expressed in astrocytes and contribute to the degradation of
monomeric Aβ species(37).
Astrocytes
also express matrix metalloproteinases MMP-2 and MMP-9 which degrade both
fibrillar and monomeric Aβ (37) (Figure 2).
Clearance
of Aβ can be mediated by extracellular proteins APOE, ApoJ/Clusterin,
β1-antichymotrypsin (ACT), and β-2-macroglobulin (β-2-M), all produced by
astrocytes (Figure 2); these proteins promote the transport of
β-2-macroglobulin Aβ across the BBB to the circulation either alone or in
association with LRP1 and VLDLR receptors (37).
Recent studies report that iPSC-derived human
astrocytes and mouse astrocytes expressing APOE4 are less efficient in clearing
Aβ than those expressing APOE3 (38). Expression of APOE4 also leads to
the degeneration of pericytes thus facilitating the breakdown of the BBB
further contributing to cognitive impairment in APOE4 carriers (39). In AD, reactive astrocytes
interact with neurons, microglia, and oligodendrocytes by releasing
feed-forward signals and contributing to the vicious cycle that leads to
neurodegeneration. Of note, β-2-macroglobulin β-2-macroglobulin
Aβ can activate the NF- κ B pathway in astrocytes, which leads to the release
of the complement protein C3 (Figure 2). The C3 binding to the microglial
receptor C3aR alters β-2-macroglobulin -amyloid beta phagocytosis while the C3
binding to the neuronal receptor C3aR disrupts dendritic morphology and network
function, both effects contributing to AD pathogenesis (40). Both NF- κB and C3 cascades are
activated in the human AD brain and AD mouse models (41). About 60% of the astrocytes in the
prefrontal cortex of AD patients are C3-expressing astrocytes (41) and could contribute to neuronal
damage; although further analyses are needed for confirmation.
In
AD, reactive astrocytes participate in shifting the excitation-inhibition
balance through secretions of GABA. In a healthy brain, astrocytes do not
contribute much to GABA production, however, in AD GABA starts to be
synthesized by astrocytes through the putrescine-MAO-B pathway (42). In this way, reactive astrocytes
start to secrete GABA thus increasing inhibition, likely to be a defensive
response against neuronal hyperexcitability that seems to be a universal result
of AD progression (43).
An increase in MAO-B expression in astrocytes,
which accompanies AD, also results in a hyperproduction of hydrogen peroxide
that may instigate neuronal damage and death (44) metabolic deficits (45) and mitochondrial dysfunction also
contribute to AD progression (46). Extensive transcriptomics and
proteomics studies revealed deficient mitochondrial bioenergetics in AD brains (47). Exposure of mouse astrocytes to Aβ
up-regulates superoxide dismutase thus increasing oxidative stress (48); while the continuous infusion of
Aβ into mice brains results in a substantial increase in the production of
hydrogen peroxide (49) overproducing astrocytes has been
recently detected in the brains of AD model mice (44). The toxic effect of Aβ on
astrocytes is manifested by mitochondrial depolarisation with subsequent loss
of Ca2+ homeostasis (50). At the same time, astrocytes can
exert neuroprotection at different stages of AD. Both astrogliosis and
microgliosis in response to Aβ increase glial
secretion of transforming growth factor (TGF-β)
(Figure 2). TGF-β protects neurons from Aβ toxicity and enhances Aβ clearance
by microglia (52). Moreover, astrocytes surrounding Aβ plaques demonstrate
phagocytic activity and can phagocytose neuritic dystrophies in both mouse
models and AD patients’ brains, further suggesting the beneficial roles of
astrocytes in AD (51). These data show that astrocytes
actively contribute to the pathogenesis of AD. At the same time, many questions
remain to be addressed. What astroglial states/phenotypes are found at
different stages of AD? How do astrocyte states/phenotypes differ between brain
regions, which are known to have different vulnerabilities to AD? How do
astrocytes crosstalk with other brain cells? Are they able to promote
neurodegeneration? How do AD risk genes modulate astroglial responses in AD?
New methodologies such as RNA sequencing and spatial transcriptomics in
combination with the use of human iPSC-derived models and CRISPR-based studies
are providing a deeper understanding of how astrocytes evolve during the course
of AD.
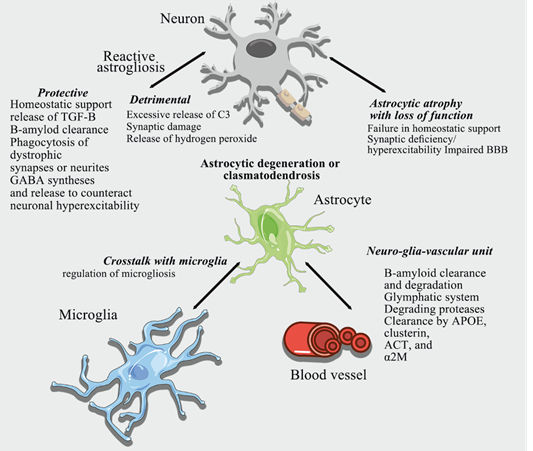
Figure 2. Contribution of astrocytes to
Alzheimer´s disease.
By
secreting neurotrophic factors such as tumor beta growth factor (TGF-β),
brain-derived neurotrophic factor (BDNF), and neuron growth factor (NGF),
astrocytes contribute to the growth of dendritic appendages and strengthen the
function of the synapse (52). They are also able to convert glucose
into lactic acid and then, neurons use this lactic acid for pyruvate synthesis
and metabolic functions (53). Astrocytes, possessing the enzyme glutamine synthetase,
receive glutamate, which is the most important neurostimulator mediator in the
CNS, and form part of the glutamine-glutamate cycle (54). Astrocyte mitochondria are
concentrated near sites of homeostatic transport (50). These mitochondria provide energy
for the Na+/K+ ATPase pump, which in turn causes the
accumulation of neurotransmitters such as glutamate and regulates cytosolic Ca2+
concentration (55). A deficiency in ATP supply may
affect glutamate clearance and increase excitotoxicity. Mitochondrial dynamics and function are also impaired in human
astrocytes with apolipoprotein E1 (APOE) allele (56).
In
addition, there are some indications that astrocytic mitochondria can be
transferred to neurons and contribute to neuronal bioenergetics. In particular, these processes seem to
support neuroprotection after stroke (57). Studies show that astrocytic
neuron transfer exerts neuroprotection in the context of Parkinson's disease (58).
Whether
this process contributes to AD remains an exciting and unanswered question.
Astrocytes seem to express lipoprotein E, neprilysin, insulin-degrading enzyme,
endothelin-converting enzyme, angiotensin-converting enzyme, and matrix
metalloproteinases, and clear Aβ peptides from nerve tissue (59). Recently the neuroprotective role
of astrocytes also was reported (23, 24). They inhibited astrocytes in the AD
model and reported that not only was cognition deficit exacerbated but also
neuroinflammation was apparent in their brain indicating the progress of AD in
the absence of astrocytes (60).
However,
it should be emphasized that astrocytes are a double edge sword playing both
inflammatory (A1 type) and anti-inflammatory roles (A2 type). Considering
diverse phenotypes of neurodegenerative A1 and neuroprotective A2 astrocytes,
and the
multidimensional functions of reactive astrocytes (41, 61), understanding the complete role of
reactive astrocytes remains at the beginning of its path.
In a
series of experiments, two groups of mice with certain characteristics were
mated together. The first group was mice
that had a gain-of-function mutation in the Aβ precursor protein (APP) gene and
the other group was mice that lacked the NLPR3 inflammasome (a mediator
molecule in the pathway inflammation related to receptors in astrocytes).
Newborn babies showed better spatial memory compared to parents with mutations
in APP, lower caspase 1 activity and more clearance of Aβ, and this itself can
be proof of the role of astrocytes in the worsening of AD (52, 62).
Investigations
show that Aβ peptides are connected to these cells through receptors located on
the surface of astrocytes, and then the activity of these cells is changed to
ward destruction and damage (52, 63-65).
One
of the most important receptors and signaling involved here is the advanced
glycation end products (RAGE/NF-κB) pathway, which is activated through the
binding of Aβ to the RAGE receptor (56, 65). RAGE has two isoforms: the s-RAGE
isoform, which is its soluble type, and the m-RAGE isoform, which is attached
to the membrane and can have harmful effects in certain conditions, including
bonding with Aβ (66).
The
activation of this path causes the activation of a chain of molecular
interactions in astrocytes and then in the entire nervous tissue. The nuclear
factor kappa light chain enhancer of activated B cells (NF-κB)
is a gene transcription complex that is normally inactively located in the
cytoplasm. This complex generally consists of two
parts. A regulatory part (in this case, called
IκB) and an acting part (67). The binding of Aβ to RAGE, through the classical or canonical
pathway, activates a kinase that phosphorylates the regulatory part of the
NF-κB complex (IKK for short). This kinase, in turn, phosphorylates IκB and
separates it from the complex and migrates into the cell nucleus, and promotes
the transcription process of cytokine genes with the help of certain factors.
Among these factors is bromodomain-containing protein 4 (BRD4).
This protein is one of the three members of the benign essential
tremors (BET) family. The members
of this family share a sequence of about 110 amino acids called bromodomain (12, 67-69). In total, all
these events cause the expression of specific inflammatory proteins and
cytokines, and adhesion molecules in white blood cells. And in this way, astrocytes change from a neurotrophic state to
a neurotoxic state (67).
In
the field of various human diseases, numerous animal studies have been planned.
Today, many specific animal models are used in medical research, including
models of stroke (70), heart failure (71, 72), and kidney failure (73). In the field of mechanism,
prevention, and treatment of Alzheimer's disease, many animal studies have been
used, for example, the study conducted by Nikkar et al (60) simultaneous administration of
bromodomain and histone deacetylase I inhibitors alleviates cognition deficit
in Alzheimer’s model of rats .
Among
the most important inflammatory cytokines that are secreted, all types of
interleukins (ILs) such as IL-1β, IL-6, IL-10, IL-17, IL-18, tumor necrosis
factor (TNF-α), interferons (IFNs) especially IFN-γ and chemokines such as
Monocyte chemoattractant protein (MCP) and macrophage inflammatory
protein (MIP) noted (74, 75).
The
release of these cytokines causes neutrophils and macrophages to be called,
neurons to be damaged, dendritic spines to be destroyed, and synapse
dysfunction, resulting in cognitive defects. The binding of these cytokines to
their receptors in neurons causes the activation of mediators such as protein
kinase C (PKC), caspase 1, caspase 3, p38 and pathways such as phosphoinositide
3-kinases, caspase 3 activity alone is sufficient to trigger the events leading
to neuronal apoptosis. Caspase 3 can also cause abnormal processing of tau
protein so that this protein is broken at the place of aspartate 421 root and a
product is created that accumulates faster than the natural form of tau in the
neuron and shortens the life of the neuron (76, 77).
In
addition, these cytokines can affect the 5'-UTR region of the APP gene, causing
its overexpression and eventually increasing Aβ (78).
They
can also cause the activation of beta and gamma-secretase enzymes in the path
of APP amyloidogenic processing and regularly increase the production and
secretion of Aβ (79). In response to amyloid beta,
calcineurin protein is activated in astrocytes and this causes the activation
of a transcription factor called a nuclear factor of activated
T-cells (NFAT) in this way, the production and secretion of cytokines will
increase (52). By binding to their receptors on
the surface of astrocytes, Aβ, and IL-1 can induce the production of
sphingomyelinase enzyme in astrocytes.
The substrate of this enzyme is sphingomyelin found in cell membranes, and by
breaking it down, it produces ceramide, which is a secondary messenger and
induces messages related to the death of neurons and even astrocytes themselves
(80, 81).
IL-1β increases the phosphorylation of tau protein and decreases a pre-synaptic
marker called synaptophysin through the p38-MAPK
pathway in primary culture media containing neurons and astrocytes (82).
IL-18
can affect N-methyl-D-aspartate (NMDA) receptors and thereby interfere with the
long-term potentiation (LTP) process (81).
NMDA receptors affect tau protein structure and function in different ways (81). For example, signals generated by
these receptors can activate calpains.
Calpains stimulate tau phosphorylation by affecting other kinases such as
glycogen synthase kinase, cyclin-dependent kinase 5 (CDK5),
extracellular signal-regulated kinases (ERK1), and ERK2. Calpain
activity also cleaves p35 to p25 and.p35 normally forms a CDK5/p35 complex with
cyclin-dependent kinase 5 (CDK5) and this complex phosphorylates tau protein to
its normal level. but p25 aggravates this process and tau
hyperphosphorylation (81, 83, 84). The research
of Farman and his colleagues showed that in APP/PS1 mice, by using the
Vorarlberg Institute for Vascular Investigation and Treatment (VIVIT) peptide,
which is an interfering factor in the Calcineurin/NFAT2 pathway, it
is possible to reduce the activity of astrocytes as well as the level of Aβ,
and the function of synapses and indicators (62). Improve learning and memory (59).
Garwood
and his colleagues concluded experiments that using the antibiotic minocycline
can prevent the activity of astrocytes and prevent the activation of caspase 3
in neurons and the production of h-tau. Additionally, they were able to
demonstrate that adding Aβ to culture media containing both neurons and
astrocytes induced neuronal death more rapidly than media containing only
neurons. In this way, they clarified the role of astrocytes and inflammation in
Alzheimer's pathogenesis (63). In 2004, Bergamaschini and colleagues showed that the
use of enoxaparin (a type of low molecular weight heparin) in Alzheimer's mice
reduced the number of active astrocytes surrounding amyloid plaques and slowed
the progression of the disease (85). Henka and his colleagues showed that the use of
pioglitazone and ibuprofen reduces inflammation in glial cells and also reduces
the amount of Aβ1-42 in APPV717I transgenic mice (86).
Medeiros
and his colleagues showed that the long-term use of IL-1 receptor-blocking
antibodies in 3xTg Alzheimer's mice improves cognitive deficits, reduces the
damage caused by tau protein, and reduces certain types of Aβ filamentous and
oligomeric peptides (87).
In
2017, Yi and his colleagues showed that Boldin, which is extracted from the
boldo tree, is effective in improving the condition of Alzheimer's mice by
inhibiting the activity of connexins in glial cells, including astrocytes (88).
In
2015, Zhang and colleagues showed that the use of paeoniflorin as an
anti-inflammatory in Alzheimer's APP/PS1 mice reduced the activity of glycogen
synthase kinase and it also prevents the chain of inflammatory processes in the
NF-κB pathway and excessive activation of astrocytes (68). Fragoulis and his colleagues
showed that the use of methysticin, an activator of the Nuclear factor
E2-related factor 2 (Nrf2) pathway (which is an anti-inflammatory transcription
factor), in the form of oral gavage during 6 months with a weekly dose of
APP/Psen1 Alzheimer's mice, it reduces astrogliosis, inflammatory cytokines
secretion and reduces long-term memory disorders (89).
In
2018, Wilkanik and his colleagues showed that intraperitoneal injection of
roscovitine in Alzheimer's mice prevented CDK5 activity and the process of
inflammatory responses (90). Astrocytes, when associated with
senile plaques, react with morphological hypertrophy manifested by thickening
processes and increased expression of the intermediate filament proteins glial
fibrillary acidic protein (GFAP), vimentin, and nestin (87).
These
data show that astrocytes are actively involved in the pathogenesis of AD. At the same time, many questions remain to
be addressed. What are the astroglial states/phenotypes
in different stages of AD? How do astrocytic states/phenotypes differ between
brain regions with different vulnerabilities to AD? How do astrocytes communicate with other brain cells? Are they
able to detect neurodegenerative disorders?
How do AD risk genes modulate astroglial responses in AD?
It
is hoped that new methods such as RNA sequencing and spatiotemporal
transcription, in combination with human induced pluripotent stem cells
(iPSC)-derived models and clustered regularly interspaced short
palindromic repeats (CRISPR) based studies, will provide a deeper understanding
of how astrocytes evolve during AD (Table 1).
Table 1. Summary of the research conducted on the role of astrocytes and
inflammatory mediators
in the development of Alzheimer's disease).
|
Results |
Type of Study |
Reference |
Year |
The name of the scholar |
|
The activity of astrocytes
and the level of amyloid beta decreased, and the function of synapses and
learning and memory indicators improved. |
Using the VIVIT
peptide Calcineurin/NFAT
pathway interfering factor) in APP/PS1 mice. |
(62) |
2012 |
Furman et al |
|
It prevented the activity of astrocytes and prevented
the activation of caspase 3 in neurons and the production of
hyperphosphorylated tau. |
Conducting tests using the antibiotic minocycline. |
(63) |
2011 |
Garwood et al |
|
Amyloid beta-induced neuronal death more quickly
and revealed the role of astrocytes and inflammation in Alzheimer's
pathogenesis. |
Comparison of the
addition of amyloid beta to vessel media containing neurons and astrocytes
with media containing only neurons. |
(63) |
2011 |
Garwood et al |
|
Reducing the number of active astrocytes surrounding
amyloid plaques and reducing the speed of disease progression |
Application of Enoxaparin (a type of low molecular
weight heparin) in Alzheimer's rats. |
(85) |
2004 |
Bergamaschini et al |
|
Reduction of
inflammation in glial cells and reduction of Aβ1-42 in APPV717I transgenic
mice. |
Pioglitazone (PPARγ
agonist) and ibuprofen were used. |
(86) |
2005 |
Heneka et al |
|
Reducing the activity of astrocytes and preventing
the activation of caspase 3 in neurons and the production of h-tau protein (hyperphosphorylated
tau( |
Use of minocycline antibiotic in h-tau mice |
(63) |
2011 |
Garwood et al |
|
Improvement of
cognitive deficits, reduction of damage caused by tau protein, and relative
reduction of certain types of amyloid beta filamentous and oligomeric
peptides. |
Long-term use of IL-1
receptor blocking antibody in 3xTg Alzheimer's mice. |
(87) |
2011 |
Medeiros et al |
|
It reduced the activity of astrocytes as well as the level of
amyloid beta and improved the function of synapses and memory. |
Using the peptide VIVIT, an interfering agent in the
Calcineurin/NFAT pathway in APP/PS1 mice. |
(62) |
2012 |
Furman et al |
|
Preventing the
activity of glycogen synthase kinase enzyme as well as the chain of
inflammatory processes in the path of NF-κB and excessive activation of
astrocytes. |
Using Paeoniflorin as
an anti-inflammatory in APP/PS1 Alzheimer's mice |
(81) |
2015 |
Zhang et al |
|
In improving the disease condition in Alzheimer's
mice. |
Preventing the activity of connexins of glial cells
and including astrocytes with the help of Boldine, which was obtained from
the Boldo tree. |
(88) |
2017 |
Yi et al |
|
It reduced astrogliosis,
reduced the release of inflammatory cytokines, and reduced long-term memory
disorders. |
Using methysticin by
oral gavage for 6 months with a dose of once a week in APP/Psen1 Alzheimer's
mice. |
(89) |
2017 |
Fragoulis et al |
|
It prevents the activity of CDK5 (cyclin-dependent
kinase 5) and the process of inflammatory responses |
Intraperitoneal injection of Roscovitine in
Alzheimer's rats. |
(90) |
2018 |
Wilkaniec et al |
|
Inhibition of astrocytes metabolism by fluorocitrate impaired
spatial memory and reduced CREB/PSD95/synaptophysin levels in the hippocampus |
Chronic co-inhibition of astrocytes metabolism (with
fluorocitrate) and also BRD4 (with JQ1) on cognition deficit at early stages
of AD in rats. |
(60) |
2022 |
Nikkar et al |
Conclusions
Astrocytes have multiple functions
in the brain and are essential for protecting neurons and maintaining
homeostasis. However, under
different pathological conditions including AD, they are associated with loss
of function associated with neuroinflammation and neurodegeneration. A thorough characterization of these cellular states, together
with morphological and functional analyses, will enhance the understanding of
how astrocytes evolve in pathology. Soon, using selective inhibitors for A1 or A2 types of astrocytes,
we may be able to correlate different astroglial states with specific stages of
Alzheimer's disease and clarify the exact role of these cells in various stages
of AD.
Author contribution
All the authors met the standard writing criteria based on the
recommendations of the International Committee of Medical Journal Editors and
all contributed equally to the writing of the work.
Conflict of interest
The authors hereby declare that there is no conflict of interest
regarding the present research.
Acknowledgments
All the authors of this review article are thanked and appreciated.
References
1. Murli S. Can Hypertension Treatment Slow
Down Alzheimer’s Disease From Progressing? hypertension. 2020.
2. Nisbet RM, Polanco J-C, Ittner LM, Götz J.
Tau aggregation and its interplay with amyloid-β. Acta Neuropathol.
2015;129(2):207-20.
3. Cacquevel M, Lebeurrier N, Cheenne S,
Vivien D. Cytokines in neuroinflammation and Alzheimer's disease. Curr. Drug
Targets. 2004;5(6):529-34.
4. Bertram L, Lill CM, Tanzi RE. The genetics
of Alzheimer disease: back to the future. Neuron. 2010;68(2):270-81.
5. Chen G-f, Xu T-h, Yan Y, Zhou Y-r, Jiang Y,
Melcher K, et al. Amyloid beta: structure, biology and structure-based
therapeutic development. Acta Pharmacol. Sin. 2017;38(9):1205-35.
6. Masters CL, Simms G, Weinman NA, Multhaup
G, McDonald BL, Beyreuther KJPotNAoS. Amyloid plaque core protein in Alzheimer
disease and Down syndrome. proc. natl. acad. sci. 1985;82(12):4245-9.
7. Iwata N, Higuchi M, Saido TCJP, therapeutics.
Metabolism of amyloid-β peptide and Alzheimer's disease. Pharmacol. Ther.
2005;108(2):129-48.
8. González-Reyes RE, Nava-Mesa MO,
Vargas-Sánchez K, Ariza-Salamanca D, Mora-Muñoz LJFimn. Involvement of
astrocytes in Alzheimer’s disease from a neuroinflammatory and oxidative stress
perspective. Front. Mol. Neurosci. 2017;10:427.
9. Fox NC, Schott JM. Imaging cerebral
atrophy: normal ageing to Alzheimer's disease. The Lancet.
2004;363(9406):392-4.
10. Mehta D, Jackson R, Paul G, Shi J, Sabbagh M.
Why do trials for Alzheimer’s disease drugs keep failing? A discontinued drug
perspective for 2010-2015. Expert Opin. Investig. Drugs. 2017;26(6):735-9.
11. Luca A, Calandra C, Luca M. Molecular bases
of Alzheimer’s disease and neurodegeneration: the role of neuroglia. Aging Dis.
2018;9(6):1134.
12. González-Reyes RE, Nava-Mesa MO,
Vargas-Sánchez K, Ariza-Salamanca D, Mora-Muñoz L. Involvement of astrocytes in
Alzheimer’s disease from a neuroinflammatory and oxidative stress perspective.
Front. Mol. Neurosci. 2017;10:427.
13. Frost GR, Li Y-M. The role of astrocytes in
amyloid production and Alzheimer's disease. Open Biol. 2017;7(12):170228.
14. Sharma HS, Castellani RJ, Smith MA, Sharma
AJIRN. The blood-brain barrier in Alzheimer's disease: novel therapeutic
targets and nanodrug delivery. Int. Rev. Neurobiol. 2012;102:47-90.
15. Ueno M, Chiba Y, Matsumoto K, Nakagawa T,
Miyanaka HJCmc. Clearance of beta-amyloid in the brain. Curr. Med. Chem.
2014;21(35):4085-90.
16. Provias J, Jeynes BJIjoasd. The role of the
blood-brain barrier in the pathogenesis of senile plaques in Alzheimer’s
disease. J. Alzheimer's Dis.2014;2014.
17. Garwood C, Ratcliffe L, Simpson J, Heath P,
Ince P, Wharton SJN, et al. astrocytes in Alzheimer's disease and other
age‐associated dementias: a supporting player with a central role. Neuropathol.
Appl. Neurobiol. 2017;43(4):281-98.
18. Wakabayashi K, Miki YJB, Shinpo NSKn.
Deposition and clearance of β-amyloid in the brain. Brain Nerve.
2013;65(12):1433-44.
19. Cai Z, Wan C-Q, Liu ZJJon. Astrocyte and
Alzheimer’s disease. J. eurol. 2017;264:2068-74.
20. Selkoe DJ, Hardy JJEmm. The amyloid
hypothesis of Alzheimer's disease at 25 years. EMBO Mol Med. 2016;8(6):595-608.
21. Verkhratsky A, Zorec R, Rodríguez JJ, Parpura
VJCoip. Astroglia dynamics in ageing and Alzheimer's disease. Curr Opin
Pharmacol. 2016;26:74-9.
22. Arranz AM, De Strooper BJTLN. The role of
astroglia in Alzheimer's disease: pathophysiology and clinical implications.
2019;18(4):406-14.
23. Yu J-T, Tan L, Hardy JJAron. Apolipoprotein E
in Alzheimer's disease: an update. Annu. Rev. Neurosci. 2014;37:79-100.
24. Verghese PB, Castellano JM, Garai K, Wang Y,
Jiang H, Shah A, et al. ApoE influences amyloid-β (Aβ) clearance despite
minimal apoE/Aβ association in physiological conditions. J. Biol. Sci.
2013;110(19):E1807-E16.
25. Rodriguez-Vieitez E, Saint-Aubert L, Carter
SF, Almkvist O, Farid K, Schöll M, et al. Diverging longitudinal changes in
astrocytosis and amyloid PET in autosomal dominant Alzheimer’s disease. Brain. 2016;139(3):922-36.
26. Serrano-Pozo A, Muzikansky A, Gómez-Isla T,
Growdon JH, Betensky RA, Frosch MP, et al. Differential relationships of
reactive astrocytes and microglia to fibrillar amyloid deposits in Alzheimer
disease. J. Neuropathol. Exp. Neurol. 2013;72(6):462-71.
27. Escartin C, Galea E, Lakatos A, O’Callaghan
JP, Petzold GC, Serrano-Pozo A, et al. Reactive astrocyte nomenclature,
definitions, and future directions. Nat. Neurosci. 2021;24(3):312-25.
28. Verkhratsky A, Zorec R, Parpura VJBP. Stratification
of astrocytes in healthy and diseased brain. Brain Pathol. 2017;27(5):629-44.
29. Alberdi E, Wyssenbach A, Alberdi M,
Sánchez‐Gómez MV, Cavaliere F, Rodríguez JJ, et al. Ca2+‐dependent endoplasmic
reticulum stress correlates with astrogliosis in oligomeric amyloid β‐treated
astrocytes and in a model of Alzheimer's disease. Aging Cell.
2013;12(2):292-302.
30. Kraft AW, Hu X, Yoon H, Yan P, Xiao Q, Wang
Y, et al. Attenuating astrocyte activation accelerates plaque pathogenesis in
APP/PS1 mice. FASEB J. 2013;27(1):187.
31. Agulhon C, Sun M-Y, Murphy T, Myers T,
Lauderdale K, Fiacco TAJFip. Calcium signaling and gliotransmission in normal
vs. reactive astrocytes. Front. Pharmacol. 2012;3:139.
32. Colombo J, Quinn B, Puissant VJBrb.
Disruption of astroglial interlaminar processes in Alzheimer’s disease. Brain
Res. Bull. 2002;58(2):235-42.
33. Yeh C-Y, Vadhwana B, Verkhratsky A, Rodríguez
JJJAn. Early astrocytic atrophy in the entorhinal cortex of a triple transgenic
animal model of Alzheimer's disease. ASN NEURO. 2011;3(5):AN20110025.
34. Jones VC, Atkinson-Dell R, Verkhratsky A,
Mohamet LJCd, disease. Aberrant iPSC-derived human astrocytes in Alzheimer's
disease. Cell Death Dis. 2017;8(3):e2696-e.
35. Chen A, Akinyemi RO, Hase Y, Firbank MJ,
Ndung’u MN, Foster V, et al. Frontal white matter hyperintensities,
clasmatodendrosis and gliovascular abnormalities in ageing and post-stroke
dementia. Brain. 2016;139(1):242-58.
36. Frost GR, Li Y-MJOb. The role of astrocytes
in amyloid production and Alzheimer's disease. Open Biol. 2017;7(12):170228.
37. Iliff JJ, Wang M, Liao Y, Plogg BA, Peng W,
Gundersen GA, et al. A paravascular pathway facilitates CSF flow through the
brain parenchyma and the clearance of interstitial solutes, including amyloid
β. Sci. Transl. Med.2012;4(147):147ra11-ra11.
38. Ries M, Sastre MJFian. Mechanisms of Aβ
clearance and degradation by glial cells. Front. Aging Neurosci. 2016;8:160.
39. Lin Y-T, Seo J, Gao F, Feldman HM, Wen H-L,
Penney J, et al. APOE4 causes widespread molecular and cellular alterations
associated with Alzheimer’s disease phenotypes in human iPSC-derived brain cell
types. Neuron. 2018;98(6):1141-54. e7.
40. Montagne A, Nation DA, Sagare AP, Barisano G,
Sweeney MD, Chakhoyan A, et al. APOE4 leads to blood–brain barrier dysfunction
predicting cognitive decline. Nature.
2020;581(7806):71-6.
41. Lian H, Zheng HJJon. Signaling pathways
regulating neuron–glia interaction and their implications in Alzheimer's
disease. J. Neurochem. 2016;136(3):475-91.
42. Liddelow SA, Guttenplan KA, Clarke LE,
Bennett FC, Bohlen CJ, Schirmer L, et al. Neurotoxic reactive astrocytes are
induced by activated microglia. Nature. 2017;541(7638):481-7.
43. Jo S, Yarishkin O, Hwang YJ, Chun YE, Park M,
Woo DH, et al. GABA from reactive astrocytes impairs memory in mouse models of
Alzheimer's disease. Nat. Med. 2014;20(8):886-96.
44. Garaschuk O, Verkhratsky AJA. GABAergic
astrocytes in Alzheimer’s disease. Aging.
2019;11(6):1602.
45. Chun H, Im H, Kang YJ, Kim Y, Shin JH, Won W,
et al. Severe reactive astrocytes precipitate pathological hallmarks of
Alzheimer’s disease via H2O2− production. Nat. Neurosci. 2020;23(12):1555-66.
46. Kapogiannis D, Mattson MPJTLN. Disrupted
energy metabolism and neuronal circuit dysfunction in cognitive impairment and
Alzheimer's disease. Lancet Neurol. 2011;10(2):187-98.
47. Wang W, Zhao F, Ma X, Perry G, Zhu XJMN.
Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: Recent
advances. Mol. Neurodegener. 2020;15:1-22.
48. Adav SS, Park JE, Sze SKJMb. Quantitative
profiling brain proteomes revealed mitochondrial dysfunction in Alzheimer’s
disease. Mol. Brain. 2019;12(1):1-12.
49. Sarkar P, Zaja I, Bienengraeber M, Rarick KR,
Terashvili M, Canfield S, et al. Epoxyeicosatrienoic acids pretreatment improves
amyloid β-induced mitochondrial dysfunction in cultured rat hippocampal
astrocytes. Am J Physiol Heart Circ Physiol. 2014;306(4):H475-H84.
50. Kaminsky YG, Kosenko EAJFRR. Effects of
amyloid-beta peptides on hydrogen peroxide-metabolizing enzymes in rat brain in
vivo. Free Radic. Res.
2008;42(6):564-73.
51. Jackson JG, O'Donnell JC, Takano H, Coulter
DA, Robinson MBJJoN. Neuronal activity and glutamate uptake decrease
mitochondrial mobility in astrocytes and position mitochondria near glutamate
transporters. J. Neurosci. Res. 2014;34(5):1613-24.
52. Gomez‐Arboledas A, Davila JC, Sanchez‐Mejias
E, Navarro V, Nuñez‐Diaz C, Sanchez‐Varo R, et al. Phagocytic clearance of
presynaptic dystrophies by reactive astrocytes in Alzheimer's disease. Glia.
2018;66(3):637-53.
53. Preman P, Alfonso-Triguero M, Alberdi E,
Verkhratsky A, Arranz AMJC. Astrocytes in Alzheimer’s disease: pathological
significance and molecular pathways. Cells. 2021;10(3):540.
54. Phillips EC, Croft CL, Kurbatskaya K, O’Neill
MJ, Hutton ML, Hanger DP, et al. Astrocytes and neuroinflammation in
Alzheimer's disease. Portland Press Ltd. 2014.
55. Allen NJ, Barres BA. Glia—more than just
brain glue. Nature. 2009;457(7230):675-7.
56. Rosenthal ZP, Kraft AW, Czerniewski L, Lee
J-M. Targeting Astrocytes With Viral Gene Therapy for Alzheimer’s Disease. Gene Therapy in Neurological Disorders:
Elsevier; 2018. p. 97-138.
57. Nikseresht Z, Ahangar N, Badrikoohi M, Babaei
PJBBR. Synergistic enhancing-memory effect of D-serine and RU360, a
mitochondrial calcium uniporter blocker in rat model of Alzheimer's disease. .
Behav. Brain Res. 2021;409:113307.
58. Schmukler E, Solomon S, Simonovitch S,
Goldshmit Y, Wolfson E, Michaelson DM, et al. Altered mitochondrial dynamics
and function in APOE4-expressing astrocytes. Cell Death Dis. 2020;11(7):578.
59. Hayakawa K, Esposito E, Wang X, Terasaki Y,
Liu Y, Xing C, et al. Transfer of mitochondria from astrocytes to neurons after
stroke. Nature.
2016;535(7613):551-5.
60. Morales I, Sanchez A, Puertas‐Avendaño R,
Rodriguez‐Sabate C, Perez‐Barreto A, Rodriguez MJG. Neuroglial transmitophagy
and Parkinson's disease. Glia. 2020;68(11):2277-99.
61. Ries M, Sastre M. Mechanisms of Aβ clearance
and degradation by glial cells. Front. Aging Neurosci. 2016;8:160.
62. Nikkar R, Esmaeili-Bandboni A, Badrikoohi M,
Babaei PJMBD. Effects of inhibiting astrocytes and BET/BRD4 chromatin reader on
spatial memory and synaptic proteins in rats with Alzheimer’s disease. Metab.
Brain Dis. 2022;37(4):1119-31.
63. Fujita A, Yamaguchi H, Yamasaki R, Cui Y,
Matsuoka Y, Yamada K-i, et al. Connexin 30 deficiency attenuates A2 astrocyte
responses and induces severe neurodegeneration in a 1-methyl-4-phenyl-1, 2, 3,
6-tetrahydropyridine hydrochloride Parkinson’s disease animal model. J.
Neuroinflammation. 2018;15:1-20.
64. Furman JL, Sama DM, Gant JC, Beckett TL,
Murphy MP, Bachstetter AD, et al. Targeting astrocytes ameliorates neurologic
changes in a mouse model of Alzheimer's disease. J. Neurosci. Res.
2012;32(46):16129-40.
65. Garwood C, Pooler A, Atherton J, Hanger D,
Noble W. Astrocytes are important mediators of Aβ-induced neurotoxicity and tau
phosphorylation in primary culture. Cell Death Dis. 2011;2(6):e167-e.
66. Jana A, Pahan K. Fibrillar
amyloid-β-activated human astroglia kill primary human neurons via neutral
sphingomyelinase: implications for Alzheimer's disease. J. Neurosci. Res.
2010;30(38):12676-89.
67. Farfara D, Lifshitz V, Frenkel DJJoc,
medicine m. Neuroprotective and neurotoxic properties of glial cells in the
pathogenesis of Alzheimer's disease. J. Cell. Mol. Med. 2008;12(3):762-80.
68. Cai Z, Liu N, Wang C, Qin B, Zhou Y, Xiao M,
et al. Role of RAGE in Alzheimer’s disease. Cell. Mol. Neurobiol.
2016;36(4):483-95.
69. Lawrence T. The nuclear factor NF-κB pathway
in inflammation. Cold Spring Harbor perspectives in biology. Cold Spring Harb.
2009;1(6):a001651.
70. Zhang H-R, Peng J-H, Cheng X-B, Shi B-Z,
Zhang M-Y, Xu R-X. Paeoniflorin atttenuates amyloidogenesis and the
inflammatory responses in a transgenic mouse model of Alzheimer’s disease.
Neurochem. Res. 2015;40(8):1583-92.
71. Stilling RM, Fischer AJNol, memory. The role
of histone acetylation in age-associated memory impairment and Alzheimer’s
disease. Neurobiol Learn Mem. 2011;96(1):19-26.
72. Pourmohammadi-Bejarpasi Z, Roushandeh AM,
Saberi A, Rostami MK, Toosi SMR, Jahanian-Najafabadi A, et al. Mesenchymal stem
cells-derived mitochondria transplantation mitigates I/R-induced injury,
abolishes I/R-induced apoptosis, and restores motor function in acute ischemia
stroke rat model. Brain Res. Bull. 2020;165:70-80.
73. Mokhtari B, Aboutaleb N, Nazarinia D,
Nikougoftar M, Tousi SMTR, Molazem M, et al. Comparison of the effects of
intramyocardial and intravenous injections of human mesenchymal stem cells on
cardiac regeneration after heart failure. Iran. J. Basic Med. Sci. 2020;23(7):879.
74. Tousi SMTR, Faghihi M, Nobakht M, Molazem M,
Kalantari E, Azar AD, et al. Improvement of heart failure by human amniotic
mesenchymal stromal cell transplantation in rats. J. Tehran Univ. Heart Cent.
2016;11(3):123.
75. Jabbari H, Roushandeh AM, Rostami MK,
Razavi-Toosi MT, Shokrgozar MA, Jahanian-Najafabadi A, et al. Mitochondrial
transplantation ameliorates ischemia/reperfusion-induced kidney injury in rat.
Biochim Biophys Acta Mol Basis Dis. 2020;1866(8):165809.
76. Mrak RE, Griffin WST. Interleukin-1,
neuroinflammation, and Alzheimer’s disease. Neurobiol. Aging. 2001;22(6):903-8.
77. Lee KS, Chung JH, Choi TK, Suh SY, Oh BH,
Hong CH. Peripheral cytokines and chemokines in Alzheimer’s disease. Dementia
and geriatric cognitive disorders. Dement Geriatr Cogn Disord.
2009;28(4):281-7.
78. Gamblin TC, Chen F, Zambrano A, Abraha A,
Lagalwar S, Guillozet AL, et al. Caspase cleavage of tau: linking amyloid and
neurofibrillary tangles in Alzheimer's disease. Proceedings of the national academy
of sciences. Proc. Natl. Acad. Sci. 2003;100(17):10032-7.
79. Rissman RA, Poon WW, Blurton-Jones M, Oddo S,
Torp R, Vitek MP, et al. Caspase-cleavage of tau is an early event in Alzheimer
disease tangle pathology. The Journal of clinical investigation. J. Clin.
Investig. 2004;114(1):121-30.
80. Lahiri D, Chen D, Vivien D, Ge Y-W, Greig N,
Rogers J. Role of cytokines in the gene expression of amyloid β–protein
precursor: Identification of a 5'-UTR-Binding nuclear factor and its
implications in Alzheimer's disease. J Alzheimers Dis. 2003;5(2):81-90.
81. Liao Y-F, Wang B-J, Cheng H-T, Kuo L-H, Wolfe
MS. Tumor necrosis factor-α, interleukin-1β, and interferon-γ stimulate
γ-secretase-mediated cleavage of amyloid precursor protein through a
JNK-dependent MAPK pathway. J. Biol. Chem. 2004;279(47):49523-32.
82. Pettus BJ, Chalfant CE, Hannun YA. Ceramide
in apoptosis: an overview and current perspectives. Biochimica et Biophysica
Acta (BBA)- Biochim Biophys Acta Mol Cell Biol Lipids. 2002;1585(2-3):114-25.
83. Zhang Y, Li P, Feng J, Wu M. Dysfunction of
NMDA receptors in Alzheimer’s disease. Neurol. Sci. 2016;37(7):1039-47.
84. Li Y, Liu L, Barger SW, Griffin WST.
Interleukin-1 mediates pathological effects of microglia on tau phosphorylation
and on synaptophysin synthesis in cortical neurons through a p38-MAPK pathway.
J. Neurosci. 2003;23(5):1605-11.
85. McBrayer M, Nixon RA. Lysosome and calcium
dysregulation in Alzheimer's disease: partners in crime. Biochem. Soc. Trans.
2013;41(6):1495-502.
86. Crews L, Masliah E. Molecular mechanisms of
neurodegeneration in Alzheimer's disease. Hum. Mol. Genet. 2010;19(R1):R12-R20.
87. Bergamaschini L, Rossi E, Storini C,
Pizzimenti S, Distaso M, Perego C, et al. Peripheral treatment with enoxaparin,
a low molecular weight heparin, reduces plaques and β-amyloid accumulation in a
mouse model of Alzheimer's disease. J. Neurosci. Res. 2004;24(17):4181-6.
88. Heneka MT, Sastre M, Dumitrescu-Ozimek L,
Hanke A, Dewachter I, Kuiperi C, et al. Acute treatment with the PPARγ agonist
pioglitazone and ibuprofen reduces glial inflammation and Aβ1–42 levels in
APPV717I transgenic mice. Brain. 2005;128(6):1442-53.
89. Medeiros R, Kitazawa M, Caccamo A,
Baglietto-Vargas D, Estrada-Hernandez T, Cribbs DH, et al. Loss of Muscarinic
M1 Receptor Exacerbates Alzheimer's Disease–Like Pathology and Cognitive
Decline. Am. J. Clin. Pathol. 2011;179(2):980-91.
90. Yi C, Ezan P, Fernandez P, Schmitt J, Saez
JC, Giaume C, et al. Inhibition of glial hemichannels by boldine treatment
reduces neuronal suffering in a murine model of Alzheimer's disease. Glia.
2017;65(10):1607-25.
91. Fragoulis A, Siegl S, Fendt M, Jansen S,
Soppa U, Brandenburg L-O, et al. Oral administration of methysticin improves
cognitive deficits in a mouse model of Alzheimer's disease. Redox Biol.
2017;12:843-53.
92. Wilkaniec A, Gąssowska-Dobrowolska M,
Strawski M, Adamczyk A, Czapski GA. Inhibition of cyclin-dependent kinase 5
affects early neuroinflammatory signalling in murine model of amyloid beta
toxicity. J. Neuroinflammation. 2018;15(1):1-18.