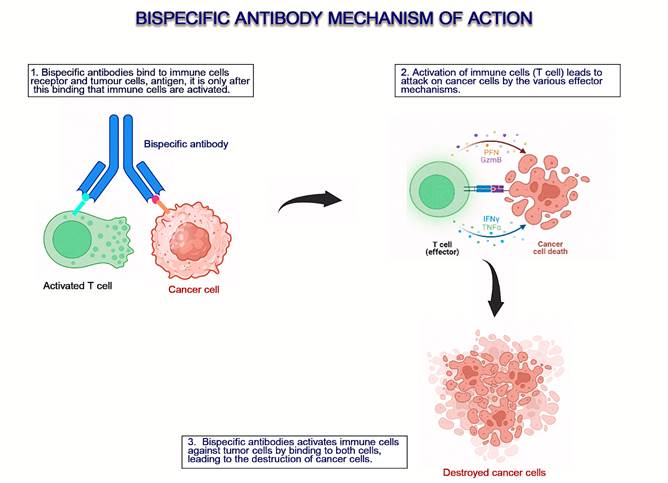
Figure 1. An overview of the mechanism of action of bispecific antibodies
(9).
Conventional
cancer immunotherapies: assessing progress and envisioning future possibilities
Paul Yiran Ntasin 1 *,
Muhammad Adamu Ibrahim 1, Ayodele Isaac Adedokun 2, Samuel
Eniola Gana 1
1 Department
of Immunology, School of Medical Laboratory Science, Usmanu
Dan Fodiyo University Sokoto, Nigeria
2 Department
of Chemical Pathology, School of Medical Laboratory Science, Usmanu Dan Fodiyo University
Sokoto, Nigeria
Corresponding Authors: Paul
Yiran Ntasin
* Email: paulyiran@gmail.com
Abstract
Cancer immunotherapy aims to modify and improve the immune system’s
fight against cancer, it is a highly promising and evolving field. It is
effective at treating a wide range of cancers, suppressing tumor growth and
improving survival rate of cancer patients. Despite the promise and fervor
around cancer immunotherapy, many challenges have limited their widespread use
and efficacy. In this review article, we consider novel cancer immunotherapies,
encouraging clinical trials and innovative strategies employed in developing
safe and effective cancer immunotherapies. It is safe to say that cancer
immunotherapy has revolutionized cancer therapy, but there are hurdles and
challenges (toxicity concerns being the most notable) that must be overcome for
safer and more effective treatment strategies. The battle against cancer is an
arduous and prolonged affair. We aim to point out what we have achieved in
recent times and outline potential strategies to mitigate our losses and chart
a course of victory.
Keywords: Cancer, Immunotherapy, Clinical trials, T-cells, Antibody
Introduction
Cancer,
normal cells in the mad pursuit of immortality, causing unprecedented mortality
and affecting so many families, remains a thorn in the flesh despite numerous
and varying onslaughts on it. The focus on cancer treatment research has
shifted from surgery, radiotherapy, and chemotherapy to immunotherapy (1). The
immune system has the ability to identify and get rid of rogue cells. Cancer
can develop when rogue cells pick up mutations that allow them to avoid the
immune system (2). These mutations enable the cancer cells to down-regulate
tumor antigen MHC I expression; suppress effector T-cells through increased
checkpoint ligands; aid regulation of immune cells through activation of
suppressor immune cells and molecules; and nurture a hostile tumor
microenvironment (3). The approval of several immunotherapies for the treatment
and management of many cancers (majorly haematological
malignancies) has generated much interest and promise in the endless possibilities cancer immunotherapies possess (1). But many
roadblocks limit their widespread adoption and efficacy in many tumors, and we
still have a long way to go. Newer strategies and modifications to cancer
immunotherapies aim to mitigate these challenges, inadvertently boosting the
immune system’s capacity to remove malignancies and improving the safety
profile of immunotherapies. Researchers are optimizing existing immunotherapies
with molecular technologies, newer sequencing tools, the evaluation of other
immune cells or molecules, and the discovery of novel tumor target antigens.
The explorative and progressive nature of scientific research ensures endless
possibilities in cancer immunotherapy. In this article, we review recently
approved cancer immunotherapies and outstanding clinical trials (CTs), their
challenges, and potential ways of optimizing cancer immunotherapy.
Antibody
Therapy and Immune Checkpoint Inhibitors (ICIs)
Antibodies
are an essential part of the body’s immune defense system. In certain
conditions, antibodies can become ineffective or insufficient, hence the
development of specific and effective antibodies in vitro (4). This has led to the emergence of
antibody diagnostics and therapeutics, which include monoclonal antibodies,
pro-antibodies, antibody-drug combinations, and bi- and tri-specific antibodies
(4). Many diseases, most notably cancer, have benefited significantly from the
use of antibody-based treatments (5). They have demonstrated success in
eliminating or suppressing many tumors, but it is not without limitations. In
this section, we consider newly approved antibody-based therapies and efforts
made to mitigate challenges encountered with this form of cancer therapy.
Monoclonal
antibody
Monoclonal
antibodies (mAbs) are antibodies that possess the
same receptor and are produced from the same B-cell line. mAbs
have found use in several immunotherapies, either in their soluble form or
bound to a membrane. Several mAbs that target
overexpressed growth factors, CD20, immune checkpoints, and CD3 have been
authorized for the treatment of cancer by the Food and Drug Administration
(FDA). Antibodies are being developed for newly discovered tumor-specific
antigens (TSA). Recently, mAbs (dinutuximab
and naxitamab) against disialoganglioside
GD2 have been approved for treating neuroblastoma (6). This has increased the
survival rate of neuroblastoma patients, but relapse has been observed in 50%
of patients (6). Monoclonal IgE antibodies that
target chondroitin sulfate proteoglycan 4 (CSPG4), which is implicated in
melanoma, induce all IgE effector functions against
melanoma in human xenograft models (7). All antibodies approved for cancer
therapy are IgG; other antibodies are now being trialed, most notably IgE, with encouraging outcomes from CTs.
Bispecific
antibodies (BsAbs)
BsAbs possesses the ability to bind to two specific antigens, which
improves its specificity. BsAb can function in
diverse ways; it helps bind immune cells to tumor cells, bind to certain
molecules to reduce their expression and block immune checkpoints (8). Clinical
BsAb can target either an antigen and CD3 in T-cells
or CD3 and immune checkpoint molecules to enhance T-cell activation (the latter
combination has demonstrated significant efficiency at T-cell activation) (9). BsAbs that target tumor antigens and CD3 molecules are
mostly used for hematological malignancies. Recently, BsAb,
mosunetuzumab-axgb, and teclistamab-cqyv
have received FDA approval for treating refractory follicular lymphoma and
refractory multiple myeloma, respectively (10,11). Mosunetuzumab
binds to CD20 on follicular lymphoma cells and CD3 on T-cells, which aids in
the destruction of the lymphoma cells (10), while Teclistamab
binds to B cell maturation antigen (BCMA) on myeloma cells and CD3 on T-cells,
leading to an effective T-cell response against myeloma cells (11). Early in
2022, the FDA granted approval for tebentafusp-tebn
usage, which binds to CD3 on T-cells and the gp100 peptide-HLA complex instead
of the tumor antigen on cancer cells, for metastatic uveal melanoma (12). Cadonilimab, a BsAb that targets
PD-1 and CTLA-4, received approval in China for relapsed or metastatic cervical
cancer (13). MEDI5752 and ABL501 are some of the BsAb
directed against immune checkpoints in clinical trials, while HLX301 targets
molecules expressed on exhausted T-cells and natural killer (NK) cells (13).
Other
combinations can be explored to fully maximize BsAb.
One of which, amivantamab-vmjw, simultaneously blocks
multiple growth factor signaling molecules to limit resistance. It has been
authorized for use in metastatic non-small cell lung cancer (NSCLC) patients
(14). Two bispecific T-cell engagers (BiTEs), epcoritamab-bysp and Glofitamab-gxbm,
received approval by the FDA in 2023 for refractory diffuse large B-cell
lymphoma (DLBCL) and high-grade B-cell lymphoma patients. They bind to CD3 on
T-cells and CD20 on lymphoma cells or healthy B-cells, leading to the
activation of T-cells and subsequent destruction of these cells (15). Pasotuxizumab, a BiTE antibody
that binds to both prostate-specific membrane antigen (PSMA) on prostate cancer
cells and the T-cell receptor (TCR) CD3, showed promise during CTs in reducing
the tumor. It is being modified and clinically tested to overcome the short
half-life and neutralize Abs against it (16) (Figure 1).
Figure 1. An overview of the mechanism of action of bispecific antibodies
(9).
Antibody-drug conjugates (ADC)
ADC
are cytotoxic agents conjugated with tumor antigen-specific antibodies, leading
to destructive effects on targeted tumor cells (17,18). About 10 ADCs have
received FDA approval for mostly hematological cancers, while about 80 ADCs are
in development (19). The major challenge with ADCs is the tendency to attack
body cells expressing the target antigen. This challenge is being addressed by
extensive screening of the target antigen, unmasking of paratopes in tumors by
TME enzymes, targeting antigens exclusively located in the TME, and adoption of
novel TSA (20). The following ADC modifications are actively being explored to
increase its efficiency: target antigen choice, chemistry of the linkers,
cytotoxic agents with greater efficiency, enhancements of conjugation
techniques, and better ADC internalization (5). In October 2022, the FDA
approved Elahere, an ADC that targets folate receptor
alpha (FRα) to treat ovarian, fallopian tube, and peritoneal cancers that are
resistant to platinum chemotherapy and express FRα (21). Sacituzumab govitecan, containing anti-Trophoblast cell-surface antigen
(TROP-2) Ab and the antineoplastic drug SN-38, has been recently authorized for
triple-negative breast cancer (TNBC) and metastatic hormone receptor (HR)+,
human epidermal growth factor 2 (HER2)-negative breast cancer (22,23).
Sacituzumab govitecan is highly effective at
targeting cancer cells and releasing its toxic payload (23).
Immune
checkpoint inhibitors (ICIs)
Immune
checkpoints regulate the immune system to protect against an uncontrolled
immune response. Cancer uses this mechanism of regulation to prevent an immune
attack on it (5). FDA authorized mAbs are available
that block immune checkpoints (most notably programmed cell death 1 ligand 1
(PD-L1) (atezolizumab, avelumab, and durvalumab), programmed cell death protein
1 (PD-1) (pembrolizumab, nivolumab, and cemiplimab),
and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) (ipilimumab, tremelimumab)), thereby allowing immune cells to attack
cancerous cells (24,25). These approved antibodies have also been combined for
higher efficacy, but only FDA-approved combinations are available for use. Poor
lymphocyte tumor infiltration and T-cell activation are some of the challenges
faced with this approach; coupling the anti-PD-L1 mAbs
with photothermal agents has proven to be effective in overcoming this
challenge (26). The FDA authorized the combo drug Opdualag
in March 2022, a combination of two ICI antibodies, nivolumab and relatlimab-rmbw, which block PD-L1 and lymphocyte
activation gene 3 (LAG3) activity, respectively. It is used for metastatic
melanoma (27). CD24 is another immune checkpoint target; it is involved in B
and T-cell regulation (28,29), cell migration (30), inhibition of phagocytosis,
and crucially contributes to the development of tumors (31). Overexpression of
CD24 has been observed in many cancers (31). mAbs
targeting CD24 have been approved for use; ALB9, G7, and SWA11 mAbs have all limited various types of cancer growth and
metastases (32,33). They have also been used in conjunction with chemotherapy
and other immunotherapies. Docosahexaenoic acid (DHA) also reduces the
manifestation of PD-L1 in cancer cells by degrading the PD-L1 ubiquitin-proteasome
and promoting C5N5-dependent PD-L1 degradation, resulting in reduced
PD-L1-mediated immunosuppression in tumor models (34). Recently, another immune
checkpoint target, Galectin-9 (an immunosuppressive regulator), has also been
targeted in CTs. Inhibition by mAbs is done in
conjunction with Ataxia telangiectasia mutated (ATM) inhibition, which leads to
remarkably suppressed tumor growth in mouse (35). Triggering receptors
expressed on myeloid cells 2 (TREM2) on monocyte-derived macrophages have been
shown to cause NK cell dysfunction in lung cancer. mAb
TREM2 inhibtion coupled with an NK cell stimulator
restores antitumor immunity in mice (36). mAbs
against some innate immune checkpoints, co-inhibitory molecules, and
co-stimulatory agonists have also been explored, but toxicity issues have
hampered progress (37,38).
Antibody-based
therapies are stable, specific to the target protein, and can induce
antibody-dependent T-cellular cytotoxicity (ADCC) by innate immune cells
(39,40). The challenges encountered in antibody-related cancer therapy include
tumor antigen mutation, immune-related adverse effects, activation of other
growth signaling pathways by tumor cells, hostility of the tumor
microenvironment (TME), poor antibody penetration, few TSA to target, immune
checkpoints, and system toxicity due to the ubiquitous nature of the target
antigen (5,41). Antibody combination therapies with other cancer therapies,
wider screening for TSA, and effective antibody delivery systems can help
eliminate some of these challenges (5). The most common way of surmounting the
systemic toxicity challenges due to the ubiquitous nature of most target
antigens is masking the antibodies to avoid binding to normal cells. Once in
the tumor environment, the antibodies are unmasked by tumor protease, thereby
activating their therapeutic functions (42). Reduced system toxicities have
been confirmed when this strategy is used, as evidenced in anti-CTLA4 DVD-Ig
(43). Other drugs under CTs include pacmilimab for
anti-PD1-L1, CX-904, EGFRxCD3, and BMS-986249 for anti-CTLA4 (5,44). Novel
tumor-specific antigens (neoantigens) are actively being investigated for
different types of cancers in order to enable the testing and development of
immunotherapeutic solutions. Circulating tumor DNA (ctDNA),
major histocompatibility complex (MHC)-II expression on tumor cells, and gene
expression profiles (GEPs) are being considered in TNBC (45). While research is
also ongoing to develop and test mAbs and BsAb against six-transmembrane epithelial antigen of
prostate (STEAP), human carcinoembryonic antigen-related cell adhesion molecule
5 (CEACAM5) and delta-like protein 3 (DLL3) expressed in different types of
prostate cancer (46,47). Additionally, studies are being conducted to ascertain
the effectiveness and safety of inhibiting the immune checkpoint B7-H3, which
is significantly expressed in neuroblastoma, as a treatment option (48).
Preclinical and clinical studies using mAbs against
anaplastic lymphoma kinase (ALK) for neuroblastoma are already in motion (49).
The innovative and explorative research methods for improving cancer antibody
therapy are commendable and will help increase the diversity of treatment
options.
Adoptive
T-Cell Therapy
Adoptive
T-cell Therapy (ACT) uses normal or engineered T-cells to identify a particular
antigen on the tumor cells and eliminate them (50). ACT aims to expand and
equip T-cells with the necessary battle armaments to eliminate elusive cancer
cells (51). ACT has brought relief and remission to many cancer patients,
mainly those with hematological malignancies, but it has yet to find
therapeutic use in solid tumors (52). The main forms of ACTs are
tumor-infiltrating lymphocytes (TILs) therapy, engineered T-cell receptor
therapy (TCR-T), and chimeric antigen receptor (CAR)-T-cell therapy (50).
TIL
therapy
TILs
was the first ACT to be developed and adopted. It
involves the harvesting and isolation of mainly T-cells exposed to tumor
antigens from metastatic lesions, expanding them, and reinfusing them with
repeated doses of interleukin-2 (IL-2) into cancer patients (1). Steve
Rosenberg was the first to experiment with this therapy in murine models; it
was later clinically trialed on metastatic melanoma patients with encouraging
results (53). The effort to extend TIL therapy is currently being expanded to
treat other solid tumors. There are currently numerous CTs of TILs for diverse
solid cancer types (advanced breast cancer, metastatic cholangiocarcinoma,
melanoma, cervical cancer, and colorectal cancer) that have remarkable
therapeutic benefits (54). A phase I CT involving the combo of TILs, IL-2, and pembrozulimab produced an effective response in metastatic
NSCLC (55). TILs therapy has a better safety profile than other ACTs, and
unlike other ACTs, it has also shown greater potential in treating solid tumors
(1).
TILs
therapy is faced with many challenges, which are being mitigated with available
technology and novel strategies. Few TILs identify autologous tumor cells; some
TILs are dysfunctional with high expression of inhibitory molecules, while
others have low affinity for tumor sites (56). With a greater understanding of
cell composition, sequencing technologies, and the utility of gene editing, the
ability to modify and improve TILs harvesting, sorting, expansion, efficacy,
and safety has greatly improved (56). Strategies aimed at countering
immunosuppression and improving TILs function in CTs include the use of
recombinant safer IL-2, knockout of transforming growth factor-β (TGF-β)
receptor-2 in TILs using CRISPR/Cas9, and knockout of TILs negative regulators
such as cytokine-induced SH2 protein (CISH), cbl-b,
and AKT1/2 (57-62). Specific phenotypes of tumor-reactive TILs (possessing
PD-1, CD39, and CD103) are being targeted to improve tumor immune responses of
TILs (63). Studies assessing the engineering of TILs to produce IL-12 (which
improves antitumor response) in small quantities are in different phases of
CTs, to particularly determine the safety of this approach (64). Combining TILs
with oncolytic viral therapy helps attract T-cells (TILs) to the TME; two
studies have confirmed the effectiveness of this strategy in mouse models
(65,66). The efforts put into addressing the difficulties encountered in TIL
therapy are remarkable.
T-cell
Receptor Engineered T-cells
T-cell
receptor (TCR) identifies antigens attached to MHC on cells or phagocytes and
initiates T-cell effector functions. TCRs are either αβ TCRs or γδ TCRs, depending on the peptide chain combination (67).
Some TCRs are specific to certain tumor antigens; the concept of TCR-T-cell
therapy is based on transferring tumor-antigen TCR gene sequence onto other
T-cells through genetic modifications; this confers the engineered T-cells with
the capacity of targeting and eliminating cells that possess that tumor antigen
(68). These TCRs are isolated from high affinity TILs or healthy T-cells
induced with tumor antigens (67). Unlike conventional CAR-T-cells (which target
only extracellular antigens), TCR-T-cells, can also target intracellular
antigens owing to their recognition of antigens bound to MHC I/II (69). The FDA
has not granted approval for any TCR-T-cell therapy; most CTs involving TCR-T
are either in phase I/II. Fatal cross-reactivity of TCR-T cells with similar or
even dissimilar antigens, insufficient T cell persistence, a paucity of
suitable TCR-T antigens, and the hostile TME are some of the obstacles
hindering the emergence of TCR-T-approved therapies. The TCR-T-cell therapies
currently in CTs mainly target NY-ESO-1, with many encouraging outcomes (1).
Increased TCR-T-cells tumor infiltration, proliferation, and effectiveness were
observed in an advanced soft tissue sarcoma treatment in a phase I trial using
TCR-T cells in combination with a nanoparticle peptide vaccine to target
NY-ESO-1 (70). Melanoma antigens recognized by T cells-1 (MART-1), MAGE-A3,
MAGE-A4, MAGE-A10, gp100, WT1, E7, and E6 are some of the other targets being
explored in CTs (71). A new gene editing technique simultaneously swaps the
initial TCR for the new one, improving the speed of clinical TCR-T production
(72). This technique has already found application in the clinical setting
(72). TCR-T has been observed in CTs to target similar antigens or
tumor-associated antigen (TAA) on normal tissues, resulting in toxicity. A
thorough preclinical assessment of TAA and the HLA is mandatory to prevent
adverse events (71).
CAR-T-cell
Therapy
Chimeric
antigen receptors (CARs) are engineered surface receptors that target a
specific antigen. They are usually attached to T-cells, but they have also
found use in NK cells and macrophages (73). Since they were first introduced,
CARs have undergone and are still undergoing various optimizations to optimize
their safety and effectiveness. They have the ability to bind to antigens in
the absence of MHC molecules (74). Additionally, CARs blend T-cell regenerative
and effector functions and the antigen-binding capacity of mAbs
(75). CAR-T cells identify varying forms of tumor antigens (proteins,
glycolipids, and carbohydrates), unlike conventional TCRs that recognize only
peptides (73). In contrast to conventional cancer therapies like chemotherapy
and radiation, CAR-T-cell therapy is a compelling substitute because of how
specifically it attacks tumors (76). CARs possess the following main domains:
the extracellular antigen binding domain (usually a modified mAb); the hinge region (which determines the length of the
antigen binding domain and provides flexibility); the transmembrane domain
(essential for CAR stability and surface expression); and the intracellular
domain (involved in intracellular signaling and co-stimulation) (77). All four
domains are essential in determining the efficiency of CAR-T-cell therapy;
these domains are constantly optimized for greater potency and therapeutic
effect (78). The intracellular domain divides CARs into five progressive
generations, targeted at optimizing their function (79-81). The signaling
pathways, functional capabilities, effectiveness, and safety of CAR-T-cell
therapy are influenced by their composition and architecture (Figure 2).
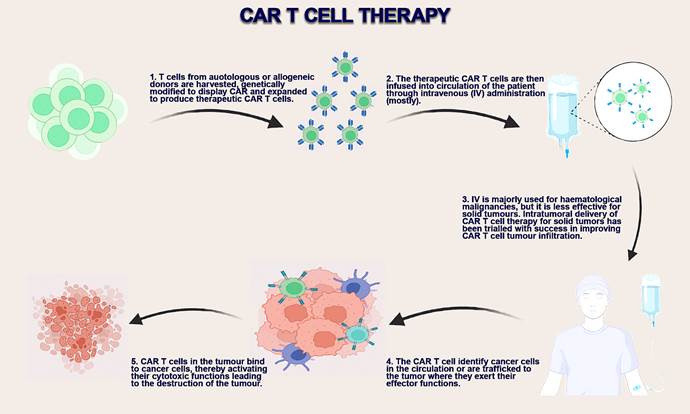
Figure 2. CAR-T-cell production, delivery methods, and mechanism of action
(75).
The
FDA has granted approval to six CAR-T-cell therapies for heamatological
cancer; these therapies are mostly used as second-line or last resort
treatments because of their high toxicity. Recent modifications have improved
the safety and effectiveness of CAR-T-cell therapies, thereby expanding their
range of use, prolonging remission, and improving survival outcomes. Ciltacabtagene autoleucel was
granted FDA approval in 2022 for usage in relapsed multiple myeloma patients
(82). The CAR-T-cell possesses two single-domain antibodies that target BCMA on
myeloma cells (82). Approved therapies are shown in Table 1. Most of these
therapies target either BCMA or CD19. Cytopenia and hypogammaglobulinaemia
are the two prominent long-term toxic effects seen in approved CAR-T-cell
therapies, while cytokine release syndrome (CRS) and neurotoxicity driven by
immune effector cells are the prominent acute toxicities (83). CTs exploring
CAR-T-cell therapies for non-haematological cancers
are burgeoning, with mixed outcomes from initial results.
Table 1. Approved CAR-T-cell therapies with
their brand name, therapeutic use and indications (82,83).
|
S/no |
CAR-T-cell therapy |
Brand name |
Therapeutic use |
Indications |
|
1 |
Idecabtagene
vicleucel |
ABECMA |
Relapsed or
refractory Multiple myeloma |
For adult
patients, to be used after four or more prior lines of therapy. |
|
2 |
Lisocabtagene
maraleucel |
BREYANZI |
Relapsed or refractory B-cell
lymphoma and follicular lymphoma |
For adult patients, to be used
after two or more lines of systemic therapy. |
|
3 |
Ciltacabtagene
autoleucel |
CARVYKTI |
Relapsed or
refractory multiple myeloma |
For patients, to
be used after four prior lines of therapy. |
|
4 |
Tisangenlecleucel |
KYMRIAH |
Relapsed or refractory B-cell acute
lymphoblastic leukemia (B-ALL) and DLBCL |
For adult patients. |
|
5 |
Brexucabtagene
autoleucel |
TECARTUS |
Relapsed or
refractory mantle cell lymphoma and B-cell precursor acute lymphoblastic
leukemia |
For adult
patients. It can be used to treat mantle cell lymphoma in other patients. |
|
6 |
Axicabtagene
ciloleucel |
YESCARTA |
Relapsed or refractory B-cell
lymphoma and Follicular lymphoma |
For patients, to be used after two
or more lines of systemic therapy. |
Widespread
adoption of CAR-T-cell therapy is restricted by some hurdles, which include
mutation or veiling of tumor antigens, effects on normal tissues that express
TAA, poor CAR-T-cell tumor invasion, hostile TME, and toxicities (84). These
issues are being addressed with innovative strategies in CTs. Studies targeting
multiple antigens have demonstrated promising efficacy, reducing the chances of
tumor antigen escape (84). CAR-T-cell extracellular ligand domains are also
being explored in preclinical and clinical studies as an alternative to
modified antibody domains to increase CAR-T-cell efficacy (85). Combination
treatment regimens with ICIs, mainly, are another vital solution to mitigating
the suppressive TME. In some studies, CAR-T-cells are genetically modified to
produce ICIs, significantly improving the efficiency of CAR-T-cell therapy
(86). Knocking out the checkpoint molecule in CAR-T-cells by CRISPR/Cas9 is
another method being explored in CTs (86). CAR-T-cells are also being
genetically modified to possess their own immunostimulatory and migratory
cytokines to resist immunosuppressive TME and improve T-cell trafficking/tumor
infiltration, respectively (84). Another method of improving CAR-T-cell tumor
infiltration is direct administration into the tumor; this has been trialed in
several studies with satisfactory outcomes (75). Non-viral vectors (mRNA and
DNA transposons systems) are being evaluated for transducing T-cells with CAR,
considering the toxicity concerns associated with viral vectors (87). Culture
expansion techniques lead to some epigenetic changes in CAR-T-cells, which
affect therapeutic outcomes. Effective expansion techniques and less
cultivation time are some of the ways to mitigate this challenge (88).
In
addition, many CTs depend on autologous T-cells as the source of CAR-T-cells
which is time-consuming and technical; the ensuing delay could be fatal for
patients with aggressive tumors (89). Chemotherapy also affects the quantity
and quality of autologous T-cells (90). Allogeneic T-cells provide large
numbers of fully operational cells and also multivalent CAR-T-cell products
(89). Despite the advantages of allogeneic CAR-T-cells, graft-versus-host
disease (GVHD) and allorejection limit their clinical
applications, but not for long (91). Eliminating the donor’s TCR with genetic
engineering can be utilized to attenuate the GVHD (91). All three adoptive
T-cell therapies share similar challenges; a breakthrough solution addressing
one of these challenges can be modified and adopted in all T-cell therapies.
Oncolytic
Virus Therapy
Oncolytic
viruses (OVs) are modified or wild viruses that infect and kill cancer cells.
They release more viruses and toxic substances that destroy cancer cells
without killing normal cells (92). Mutations in cancer cells leave them
susceptible to viral infection due to an altered antiviral defense system (93).
OVs kill infected cancer cells through toxic viral activities and numerous
immune-killing functions (94). The OV alters the cell death processes of the
tumor cells and uses the cells resources for its own survival and reproduction
before moving on to infect the next tumor cell (95). OVs also release
pathogen-associated molecular patterns (PAMPs) and death-associated molecular
patterns (DAMPs) to amplify specific antitumor immune responses or through the
effects of proteins encoded in engineered OVs (94). The OVs selected are
weakened strains or harmless viruses that are capable of infecting cancer cells
and stimulating the immune system (96).
OVs
promote inflammation in the tumor, which is an excellent way of attracting and
activating immune cells. Phagocytes engulf tumor antigens and present them to
T-cells, thereby activating T-cell antitumor activity (97). CTs on OVs or
combination therapies (especially with ICIs) are increasing due to the
therapy’s safe profile. In Japan, Teserpaturev, a
recombinant oncolytic herpes simplex virus type-1 (HSV-1), was granted
provisional regulatory approval for stereotactic intratumoral
therapy of patients with inoperable glioma (97). Approval has been granted to
four OVs in various countries, but talimogene laherparepvec (T-VEC) is the sole universally authorized OV
therapy. It received approval in 2015 for usage in recurrent melanoma patients,
but it is still being optimized and trialed for use in other cancers (98). Telomelysin (monotherapy and combinational therapy) for
head and neck cancer patients is another OV in phase II CT in the United States
of America but has been approved for use in Canada and the Asia-pacific region
(98). Telomelysin is an adenoviral OV that possesses
the human telomerase reverse transcriptase gene (hTERT) promoter, which is
prominent in cancer cells (99). Canerpaturev, a
mutant HSV-1, is another OV that awaits FDA approval; its’ efficacy at
eliciting an immune response and destroying tumor cells is well documented in
several studies (100). OVs are also engineered to act as viral vectors. Nadofaragene firadenovec-vncg (Adstiladrin), an adenoviral vector for gene therapy
(containing Interferon-α2b gene) was granted FDA approval in December 2022 for
use in non-muscle-invasive bladder cancer patients (101). Many other viral
vectors are at different stages of CTs. Genetic engineering of OVs has
increased the possibilities and potential of OVs therapy. The modifications
include the surface display of antitumor antibodies, the incorporation of
immunomodulatory genes (97) and the introduction of cell
death-inducing factors. Arakai et al. showed that Ad
OBP-702, an engineered OV expressing p53, enhanced ICD (102). Recombinant
Newcastle disease virus (NDV), NDV-MIP3α equipped with the macrophage
inflammatory protein-3α (MIP-3α) enhanced tumor killing as well as improved the
maturation and stimulation of dendritic cells (DCs) (103), 4-1BBL, a T-cell immunostimulator incorporated into the VACV/MVA vaccine,
enhanced CD8 T-cell activation and also destroyed tumor cells (104). OVs
penetrate solid tumors, which is an important advantage as it improves the
efficacy of other immunotherapies, which are usually ineffective against solid
tumors. The few challenges encountered with OVs therapy include attacks on OVs
by the immune system, safety concerns, an insufficient immune response, OV
delivery systems, and OV tumor penetration (105). These challenges would have
to be addressed before widespread adoption and clinical usage of OVs
materialize.
Cancer
Vaccine
Vaccines
are molecules or organisms that stimulate the immune system to provide
protection against a particular antigen or organism (106). In 1980, the
inaugural cancer vaccine was devised, comprising cancer cells and extracts
(106). Cancer vaccines artificially expose the immune system to cancer
antigens, thereby priming the body’s defenses against future exposure to that
antigen (107). The human papillomavirus (HPV) vaccine and the hepatitis B virus
(HBV) vaccine are the two approved prophylactic cancer vaccines. They avert HPV
and HBV infection, which are associated with an incidence of cervical and
hepatic cancer, respectively (107). Bacillus calmette-guerin
(BCG) vaccine, which is used for tuberculosis, has been approved for bladder
cancer, while Sipuleucel-T helps treat prostate
cancer (107). Both therapeutic and prophylactic vaccines have limited uses
considering the multiplicity and plasticity of cancer antigens and the fact
that the immune system they aim to stimulate is easily evaded or repressed by
tumors. Cancer vaccine research was considered a failure by some, but there has
been renewed interest in the use of neoantigens in cancer vaccines. There are
many forms of cancer vaccines (peptide-based, nucleic acid, and DC vaccines)
based on neoantigens in CTs. Peptide-based vaccines are specific,
cost-friendly, and safe, with many studies exploring its utility, one recent
study showcased the inducement of antitumor T-cell immune responses in NSCLC
models treated with personalized peptide vaccine (108). In a single-patient
study, the administration of DNAJB1-PRKACA-peptide vaccine with a
poly-ADP-ribose polymerase inhibitor induced a specific and efficient T-cell
response against DNAJB1-PRKACA, the oncogenic driver in fibrolamellar
hepatocellular carcinoma (109). There was no relapse in the patient 21 months
after vaccination (109), which is remarkable. The main challenge with peptide
vaccines is moderate immunogenicity (110). Recent studies have made headway in
solving this challenge with the conjugation of nanoparticles or
immunostimulatory adjuvants (heat shock protein 70, C-terminal of diphtheria
toxin) with the vaccine, and the results from the CTs are impressive (110).
Nucleic acid vaccines are relatively cheap; they also induce cellular and
humoral immunity, but the immune response induced is disappointing most of the
time. Personalized DNA and RNA vaccines are being explored by researchers with
modifications to improve their formulation and efficacy. The success of
SARS-CoV-2 mRNA vaccines has led to renewed interest in mRNA solutions for
cancer. Over 30 mRNA solutions are in different stages of CTs with mixed
initial outcomes (111). BNT112, encoding 5 prostate-specific antigens, recorded
positive immune outcomes in metastatic castration-resistant prostate cancer (mCRPC) from initial data in phase I/IIa
CT (112). BNT121, BNT122, and CV9201 are other mRNA vaccines inducing favorable
immunological responses in CT (113). It should be noted that a good number of
mRNA vaccines do not elicit a significant immunological response (111). As with
other cancer immunotherapy solutions, combination therapy enhances immune
responses. mRNA encoding immune costimulatory molecules, Toll-like receptor
(TLR)-4, and TAA incorporated into dendritic cells administered with
ipilimumab, a mixture identified as Trimix DC-MEL, elicited robust T-cell
responses (particularly CD8 T-cells) in melanoma patients during phase II CT
(113). DC-based vaccines induce potent immune responses but are expensive to
produce. DCs are harvested and pulsed with neoantigens. mRNA or peptides can be
incorporated into the DC and infused into cancer patients. Other DC fusion
techniques include DC-tumor fusion and electrofusion. Several mRNA-loaded DC
vaccines in CTs elicit potent antitumor T-cell immune responses with an
excellent safety profile in various cancers (114). Reduced tumor antigen
exposure through mutation or low expression, the heterogeneous nature of
tumors/tumor antigen, appropriate vaccine platforms, and insufficient immunostimulation are some of the challenges encountered in
cancer vaccine development (115). These challenges do not deter research aimed
at optimizing cancer vaccines for safe and efficient cancer therapy (Figure 3).
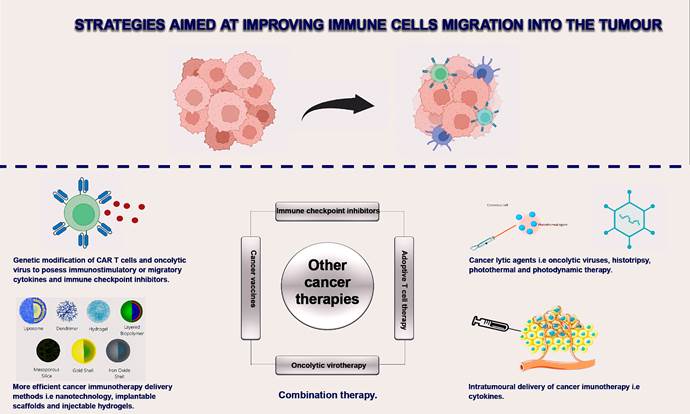
Figure 3. Improving immune cell trafficking and
migration to tumours. The hostile tumour microenvironment is one of the major challenges
limiting the efficacy of cancer immunotherapies. A summary of the various
strategies employed to mitigate these challenges is outlined in the diagram
(1,110).
Conclusions
For years now, treatment options for
cancer conditions have consistently gained much attention, with increased life
expectancy among affected patients and a better understanding of
immunosurveillance against cancer cells. Cancer immunotherapy has
revolutionized the treatment of different types of cancer conditions with
remarkable success. Several cancer immunotherapy regimens have been developed,
including adoptive T-cell therapy, immune checkpoint inhibitors, antibody
therapies, oncolytic virus therapy, and cancer vaccines, with significant breakthroughs
and concomitant increases in patient’s quality of life and survival. However,
it is important to note that patient responsiveness to immunotherapy does not
cut across all patients due to differences in human genetic composition, tumor
antigen heterogeneity, stage of the cancer, and the fitness of the immune
system. Considering this, further research is ongoing on improving the
effectiveness of immunotherapies and reducing their toxicity concerns. It is
worth noting that the next generation of cancer immunotherapies will greatly
change the status quo in the battle against cancer progression and metastasis.
Limitations of the study
1.
The study excluded other
non-conventional immunotherapies including cytokine cancer immunotherapy.
2.
Emerging technologies intended to
enhance cancer immunotherapy delivery were not covered in the study.
Author contribution
PYN wrote the antibody-based therapy section and part of the other
sections. He also reviewed the corrections and drafted the figures and table. MAI
wrote the oncolytic virus and cancer vaccine sections. AAI wrote the
conclusion and handled the plagiarism and grammar checks. GSE wrote the
introduction and adoptive cell therapy sections. All authors contributed to the
manuscript revision and approved the final draft.
Conflict of interest
All authors declare that they have no conflicts of interest.
References
1.
Liu C, Yang M, Zhang D, Chen M, Zhu D. Clinical
cancer immunotherapy: Current progress and prospects. Front Immunol. 2022; 13:
961805.
2.
Tan AC, Bagley SJ, Wen PY, et al. Systematic review
of combinations of targeted or immunotherapy in advanced solid tumors. J Immunother Cancer.
2021; 9 (7): e002459.
3.
Chang H, Qiu J, Buck MD, et al. Metabolic
competition in the tumor microenvironment is a driver
of cancer progression. Cell. 2015; 162 (6): 1229-1241.
4.
Liu L, Chen J.
Therapeutic antibodies for precise cancer immunotherapy: current and future
perspectives. Med Rev. 2023; 2 (6): 555-569.
5.
Carter PJ,
Rajpal A. Designing antibodies as therapeutics. Cell. 2022; 185: 2789–2805.
6.
Chan GC, Chan
CM. Anti-GD2 directed immunotherapy for high-risk and metastatic neuroblastoma.
Biomolecules. 2022; 12 (3): 358.
7.
Chauhan J,
Grandits M, Palhares L, et al. Anti-cancer pro-inflammatory effects of an IgE antibody targeting the melanoma-associated antigen
chondroitin sulfate proteoglycan 4. Nat Commun. 2023;
14: 2192.
8.
Ma J, Mo Y,
Tang M, et al. Bispecific Antibodies: From Research to Clinical Application.
Front Immunol. 2021; 12: 626616.
9.
Goebeler ME, Bargou RC. T-cell-engaging therapies - BiTEs
and beyond. Nat Rev Clin Oncol. 2020; 17: 418–434.
10.
Moreau P, Garfall AL, van de Donk N, et al. Teclistamab
in relapsed or refractory multiple myeloma. N Engl J Med. 2022; 387: 495–505.
11.
Budde LE,
Assouline S, Sehn LH, et al. Single-agent mosunetuzumab
shows durable complete responses in patients with relapsed or refractory B-cell
lymphomas: phase I dose escalation study. J Clin Oncol. 2022; 40: 481–491.
12.
Nathan P,
Hassel JC, Rutkowski P, et al. Overall survival benefit with tebentafusp in metastatic uveal melanoma. N Engl J Med.
2021; 385: 1196–1206.
13.
Zhang T, Lin Y,
Gao Q. Bispecific antibodies targeting immunomodulatory checkpoints for cancer
therapy. Cancer Biol Med. 2023; 20 (3): 181-195.
14.
Moores SL, Chiu ML, Bushey BS, et al. A novel
bispecific antibody targeting EGFR and cMet is
effective against EGFR inhibitor-resistant lung tumors. Cancer Res. 2016; 76:
3942–3953.
15.
Riaz R, Khan A,
Siddiqui T. Epcoritamab-bysp (Epkinly)-A
phenomenal breakthrough in the treatment of diffuse large B-cell lymphoma. Rare
Tumors. 2023; 15: 20363613231193566.
16.
Deegen P, Thomas O, Nolan-Stevaux
O, et al. The PSMA-targeting half-life extended BiTE
Therapy AMG 160 has potent antitumor activity in preclinical models of
metastatic castration-resistant prostate cancer. Clin Cancer Res. 2021; 27:
2928-2937.
17.
Tong J, Harris
PWR, Brimble MA, Kavianinia I. An insight into FDA
approved antibody-drug conjugate for cancer therapy. Molecules. 2021; 26 (19):
5847.
18.
Su D, Zhang D.
Linker design impacts antibody-drug conjugate pharmacokinetics and efficacy via
modulating the stability and payload release efficiency. Front Pharmacol. 2021; 12:
687926.
19.
Dean AQ, Luo S,
Twomey JD, Zhang B. Targeting cancer with antibody drug conjugates: promises
and challenges. mAbs. 2021; 13 (1): 1951427.
20.
Boni V, Sharma
MR, Patnaik A. The resurgence of antibody drug conjugates in cancer
therapeutics: Novel targets and payloads. Am Soc Clin Oncol Educ Book. 2020;
40: 1-17.
21.
Matulonis UA, Lorusso D, Oaknin A, et al.
Efficacy and safety of Mirvetuximab Soravtansine in patients with platinum-resistant ovarian
cancer with high folate receptor alpha expression:results
from the SORAYA study. J Clin Oncol. 2023; 41 (13): 2436-2445.
22.
Bardia A,
Hurvitz SA, Tolaney SM, et al. Sacituzumab Govitecan in Metastatic Triple-Negative Breast Cancer. N
Engl J Med. 2021; 384: 1529–1541.
23.
Rugo HS, Bardia
A, Marme F, et al. Sacituzumab Govitecan
in Hormone receptor-positive/Human epidermal growth factor 2-negative
metastatic breast cancer. J Clin Oncol. 2022; 40 (29): 3365-3376.
24.
Hargadon KM,
Johnson CE, Williams CJ. Immune checkpoint blockade therapy for cancer: an
overview of FDA approved immune checkpoint inhibitors. Int Immunopharmacol.
2018; 62: 29–39.
25.
Sharma P,
Allison JP. The future of immune checkpoint therapy. Science. 2015; 348: 56–61
26.
Huang L, Li Y,
Du Y, et al. Mild photothermal therapy potentiates anti-PD-L1 treatment for
immunologically cold tumors via an all-in-one and all-in-control strategy. Nat Commun. 2019; 10: 4871.
27.
Tawbi HA, Schadendorf D,
Lipson EJ, et al. Relatlimab and nivolumab versus
nivolumab in untreated advanced melanoma. N Engl J Med. 2022; 386 (1): 24-34.
28.
Wu H, Su Z,
Barnie PA. The role of B regulatory (B10) cells in inflammatory disorders and
their potential as therapeutic targets. Int Immunopharmacol.
2020; 78: 106111.
29.
Shi Y, Zhu J,
Liu J-Q, Talebian F, Li M, Bai X-F. CD24 is expressed
on FoxP3+ regulatory T-cells and regulates their function. Am J Transl Res. 2022; 14: 2291–2300.
30.
Runz S, Mierke
CT, Joumaa S, Behrens J, Fabry B, Altevogt P. CD24 induces localization of β1
integrin to lipid raft domains. Biochem Biophys Res Commun. 2008; 365:
35–41.
31.
Altevogt P,
Sammar M, Hüser L, Kristiansen G. Novel insights into
the function of CD24: A driving force in cancer. Int J Cancer. 2021; 148:
546–559.
32.
Bretz NP,
Salnikov AV, Perne C, et al. CD24 controls Src/STAT3
activity in human tumors. Cell Mol Life Sci. 2012; 69: 3863–3879.
33.
Chen Z, Wang T,
Tu X, Xie W, He H. Antibody-based targeting of CD24 enhances antitumor effect
of cetuximab via attenuating phosphorylation of Src/STAT3.
Biomed. Pharmacother. 2017; 90: 427–436.
34.
Zhang H, Chen
H, Yin S, et al. Docosahexaenoic acid reverses PD-L1 mediated immune
suppression by accelerating its ubiquitin-proteasome degradation. J Nutr Biochem. 2023; 112: 109186.
35.
Zheng S, Song
J, Linghu D, et al. Galectin-9 blockade synergizes
with ATM inhibition to induce potent anti-tumor immunity. Int J Biol Sci. 2023;
19 (3): 981-993.
36.
Park MD,
Reyes-Torres I, LeBerichel J, et al. TREM2
macrophages drive NK cell paucity and dysfunction in lung cancer. Nat Immunol.
2023; 24 (5): 792-801.
37.
Segal NH, Logan
TF, Hodi FS, et al. Results from an integrated safety analysis of urelumab, an agonist anti-CD137 monoclonal antibody. Clin
Cancer Res. 2017; 23: 1929–1936.
38.
Pietsch EC,
Dong J, Cardoso R, et al. Antileukemic activity and tolerability of anti-human
CD47 monoclonal antibodies. Blood Cancer J. 2017; 7 (2): e356.
39.
Roopenian DC, Akilesh S. FcRn:
the neonatal Fc receptor comes of age. Nat Rev Immunol. 2007; 7: 715–725.
40.
Nimmerjahn F, Ravetch JV. Fc
gamma receptors: old friends and new family members. Immunity 2006; 24: 19–28.
41.
Shankar B,
Zhang J, Naqash AR, et al. Multisystem immune-related
adverse events associated with immune checkpoint inhibitors for treatment of
non-small cell lung cancer. JAMA Oncol. 2020; 6: 1952–1956
42.
Vasiljeva O, Hostetter DR, Moore SJ, Winter MB. The
multifaceted roles of tumor-associated proteases and harnessing their activity
for prodrug activation. Biol Chem. 2019; 400: 965–977.
43.
Pai CS, Simons
DM, Lu X, et al. Tumorc onditional
anti-CTLA4 uncouples antitumor efficacy from immunotherapy-related toxicity. J
Clin Invest. 2019; 129: 349–363.
44.
Naing A,
Thistlethwaite F, De Vries EGE, et al. CX-072 (pacmilimab),
a Probody ((R)) PD-L1inhibitor, in advanced or
recurrent solid tumors (PROCLAIM-CX-072): an open-label dose finding and first-in-human
study. J Immunother Cancer. 2021; 9 (7): e002447.
45.
Garufi G, Carbognin L, Schettini F, et al. Updated Neoadjuvant
Treatment Landscape for Early Triple Negative Breast Cancer: Immunotherapy,
Potential Predictive Biomarkers, and Novel Agents. Cancers. 2022; 14: 4064.
46.
Lin TY, Park
JA, Long A, Guo HF, Cheung NV. Novel potent anti-STEAP1 bispecifc
antibody to redirect T-cells for cancer immunotherapy. J Immunother
Cancer. 2021; 9 (9): e003114.
47.
Dorff TB,
Narayan V, Forman SJ, Zang PD, Fraietta JA, June CH.
Novel redirected T-cell immunotherapies for advanced prostate cancer. Clin
Cancer Res. 2022; 28: 576-584.
48.
Pulido R,
Nunes-Xavier CE. Hopes on immunotherapy targeting B7-H3 in neuroblastoma. Transl Oncol. 2023; 27: 101580.
49.
Furman WL.
Monoclonal antibody therapies for high risk
neuroblastoma. Biologics 2021; 15: 205–219.
50. Feldman SA, Assadipour Y, Kriley I, Goff SL, Rosenberg SA. (2015).
Adoptive Cell Therapy—Tumor-Infiltrating Lymphocytes,
T-Cell Receptors, and Chimeric Antigen Receptors. Semin oncol.
2015; 42 (4): 626-639.
51. Alnefaie A, Albogami
S, Asiri Y, et al. Chimeric Antigen Receptor T-Cells: An Overview of Concepts,
Applications, Limitations, and Proposed Solutions. Front Bioeng
Biotechnol. 2022; 10: 797440.
52. Rohaan MW, Wilgenhof S, Haanen JBAG. Adoptive cellular therapies: the
current landscape. Virchows Arch. 2019; 474 (4):
449–461.
53. Rosenberg SA, Packard
BS, Aebersold PM, et al. Use of tumor-infiltrating
lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic
melanoma. a preliminary report. N Engl J Med. 1988; 319: 1676–1680.
54. Zhao Y, Deng J, Rao S,
et al. Tumor infiltrating lymphocyte (TIL) therapy
for solid tumor treatment: progressions and
challenges. Cancers. 2022; 14 (17): 4160.
55. Creelan BC, Wang C, Teer JK, et al. Tumor-infiltrating lymphocyte treatment for
anti-PD-1-resistant metastatic lung cancer: a phase 1 trial. Nat Med. 2021; 27
(8): 1410–1418
56.
Tasa L, Jedemaa I, Haanen
J. Novel strategies to improve efficacy of treatment with tumor-infiltrating
lymphocytes (TILs) for patients with solid cancers. Curr
Opin Oncol. 2023; 35: 107–113.
57. Fix SM, Forget MA,
Sakellariou-Thompson D, et al. CRISPR-mediated TGFBR2 knockout renders human
ovarian cancer tumor-infiltrating lymphocytes
resistant to TGF-beta signaling. J Immunother Cancer. 2022; 10: e003750.
58. Palmer DC, Webber BR,
Patel Y, et al. Internal checkpoint regulates T-cell neoantigen reactivity and
susceptibility to PD1 blockade. Med. 2022; 3 (10): 682–704.
59. Kumar J, Kumar R,
Kumar Singh A, et al. Deletion of Cbl-b inhibits
CD8(þ) T-cell exhaustion and promotes CAR-T-cell function. J Immunother Cancer. 2021; 9 (1): e001688.
60. Liu Y, Zhou N, Zhou L,
et al. IL-2 regulates tumor-reactive CD8(þ) T-cell
exhaustion by activating the aryl hydrocarbon receptor. Nat Immunol. 2021; 22:
358–369.
61. Feng H, Qiu L, Shi Z,
et al. Modulation of intracellular kinase signaling
to improve TIL stemness and function for adoptive cell therapy. Cancer Med.
2022; 12 (3): 3313-3327.
62. Deak CL, Nicolini V,
Hashimoto M, et al. PD-1-cis IL-2R agonism yields better effectors from
stem-like CD8(þ) T-cells. Nature. 2022; 610: 161–172.
63. Hanada KI, Zhao C,
Gil-Hoyos R, et al. A phenotypic signature that identifies neoantigen-reactive
T-cells in fresh human lung cancers. Cancer Cell. 2022; 40: 479–493,
64. Jones DS, Nardozzi JD,
Sackton KL, et al. Cell surface-tethered IL-12 repolarizes the tumor immune microenvironment to enhance the efficacy of
adoptive T-cell therapy. Sci Adv. 2022; 8 (17): eabi8075.
65. Ye K, Li F, Wang R, et
al. An armed oncolytic virus enhances the efficacy of tumor-infiltrating
lymphocyte therapy by converting tumors to artificial
antigen presenting cells in situ. Mol Ther. 2022; 30: 3658–3676.
66. Feist M, Zhu Z, Dai E,
et al. Oncolytic virus promotes tumor-reactive
infiltrating lymphocytes for adoptive cell therapy. Cancer Gene Ther. 2021; 28:
98–111.
67. Shah K, Al-Haidari A,
Sun J, Kazi JU. T-cell receptor (TCR) signaling in
health and disease. Signal Transduct Target Ther.
2021; 6 (1): 412.
68. Chandran SS, Klebanof CA. T-cell receptor-based cancer immunotherapy:
emerging efficacy and pathways of resistance. Immunol Rev. 2019; 290 (1):
127–147
69. Chen L, Qiao DJ, Wang
JT, Tian G, Wang MJ. Cancer immunotherapy with lymphocytes genetically
engineered with T-cell receptors for solid cancers. Immunol Lett. 2019; 216:
51–62.
70. Ishihara M, Nishida Y,
Kitano S, et al. A phase 1 trial of NY-ESO-1-specific TCR-engineered T-cell
therapy combined with a lymph node-targeting nanoparticulate peptide vaccine
for the treatment of advanced soft tissue sarcoma. Int J cancer, 2023; 152 (12):
2554–2566.
71. Shafer P, Kelly LM,
Hoyos V. Cancer therapy with TCR-engineered T-cells: current strategies,
challenges and prospects. Front Immunol. 2022; 13: 835762.
72. Zhang Y, Liu Z, Wei W,
Li Y. TCR engineered T-cells for solid tumor
immunotherapy. Exp Hematol Oncol. 2022; 11 (1): 38.
73. Abbott RC, Cross RS,
Jenkins MR. Finding the Keys to the CAR: Identifying Novel Target Antigens for
T-cell Redirection Immunotherapies. Int J Mol Sci. 2020; 21 (2): 515.
74. Lim WA, June CH. The
Principles of Engineering Immune Cells to Treat Cancer. Cell. 2017; 168:
724–740.
75. Kozani SP, Kozani
SP, Najafabadi AM, Yousefi F, Mirarefin
SM, Rahbarizadeh F. Recent Advances in solid tumor CAR-T-cell Therapy: Driving tumor
Cells from Hero to Zero? Front Immunol. 2022; 13: 795164.
76. Sadelain M, Brentjens
R, Rivière I. The Basic Principles of Chimeric Antigen Receptor Design. Cancer Discov. 2013; 3: 388–398.
77. Lindner SE, Johnson
SM, Brown CE, Wang LD. Chimeric antigen receptor signaling:
Functional consequences and design implications. Sci Adv. 2020; 6 (21):
eaaz3223.
78. Jayaraman J, Mellody
MP, Hou AJ, et al. CAR-T design: Elements and their synergistic function. EBioMedicine. 2020; 58: 102931.
79. Guedan S, Calderon H, Posey AD, Maus
MV. Engineering and Design of Chimeric Antigen Receptors. Mol Ther Methods Clin
Dev. 2019; 12: 145–156.
80. Chmielewski M, Abken H. TRUCKS, the Fourth‐generation CAR-T-cells: Current
Developments and Clinical Translation. Adv. Cell. Gene Ther. 2020; 3 (3):
1145-1154.
81. Tokarew N, Ogonek J, Endres S, von Bergwelt-Baildon M, Kobold S. Teaching an Old Dog New
Tricks: Next-Generation CAR-T-cells. Br. J. Cancer. 2019; 120: 26–37.
82. Berdeja JG, Madduri D,
Usmani SZ, et al. Ciltacabtagene autoleucel,
a B-cell maturation antigen-directed chimeric antigen receptor T-cell therapy
in patients with relapsed or refractory multiple myeloma (CARTITUDE-1): a phase
1b/2 open-label study. Lancet. 2021; 24 (398): 314-324.
83. Cappell KM,
Kochenderfer JN. Long term outcomes following CAR-T-cell therapy: what we know
so far. Nat Rev Clin Oncol. 2023; 20: 359-371.
84. Sterner RC, Sterner
RM. CAR-T-cell therapy: current limitations and potential strategies. Blood
Cancer J. 2021; 11 (4): 69.
85. Ramirez-Chacon A,
Betriu-Mendez S, Bartolo-Ibars A, Gonzalez A, Marti
M, Juan M. ligand-based CAR-T-cell: Different strategies to drive T-cells in
future new treatments. Front Immunol. 2022; 13: 932559.
86. Gumber D, Wang LD.
Improving CAR-T immunotherapy: overcoming the challenges of T-cell exhaustion. EBioMedicine 2022; 77: 103941.
87. Martinez M, Moon EK.
CAR-T-cells for solid tumors: new strategies for
finding, infiltrating and surviving in the tumor
microenvironment. Front Immunol. 2019; 10: 128.
88. Salz L, Seitz A,
Schafer D, et al. Culture expansion of CAR-T-cells results in aberrant DNA
methylation that is associated with adverse clinical outcome. Leukaemia. 2023;
37 (7): 1-11.
89. Depil S, Duchateau P, Grupp SA, Mufti
G, Poirot L. ‘Off-the-shelf’ Allogeneic CAR-T-cells: Development and
Challenges. Nat Rev Drug Discov. 2020; 19: 185–199.
90. Zhao Z, Chen Y,
Francisco NM, Zhang Y, Wu M. The Application of CAR-T-cell Therapy in Hematological Malignancies: Advantages and Challenges. Acta
Pharm Sin B. 2018; 8: 539–551.
91. Martínez-Bedoya D,
Dutoit V, Migliorini D. Allogeneic CAR-T-cells: An Alternative to Overcome
Challenges of CAR-T-cell Therapy in Glioblastoma. Front Immunol. 2021; 3 (12):
506.
92. Jennings VA, Scott GB,
Rose AMS, et al. (2019). Potentiating Oncolytic Virus-Induced Immune-Mediated Tumor Cell Killing Using Histone Deacetylase Inhibition.
Mol. Ther. 2019; 27 (6): 1139–1152.
93. Harrington K, Freeman
DJ, Kelly B, Harper J, Soria JC. Optimizing Oncolytic Virotherapy in Cancer
Treatment. Nat. Rev. Drug Discov. 2019; 18 (9):
689–706.
94. Russell SJ, Peng KW,
Bell J. Oncolyticytic virotherapy. Nat Biotechnol. 2012;30(7):658-670.
95. Cattaneo R, Miest T,
Shashkova EV, Barry MA. Reprogrammed viruses as cancer therapeutics: targeted,
armed and shielded. Nat Rev Microbiol. 2008; 6:
529–540.
96. Mondal M, Guo J, He P,
Zhou D. Recent advances of oncolytic virus in cancer therapy. Hum Vaccin Immunother. 2020; 16 (10):
2389-2402.
97. Russell SJ, Bell JC,
Engeland CE. et al. Advances in oncolytic virotherapy. Commun
Med. 2022; 2: 33.
98. Shalhout SZ, Miller DM, Emerick KS,
Kaufman HL. Therapy with oncolytic viruses: Progess
and challenges. Nat Rev Clin Oncol. 2023; 20: 160-177.
99.
Shah MA, Eads
JR, Sarkar S, et al. Phase II study of telomelysin
(OBP-310) in combination with pembrolizumab in gastroesophageal (GEA)
adenocarcinoma. J Clin Oncol. 2023; 41 (16): 4052.
100. Eissa IR, Mukoyama N, Abdelmoneim M, et al. oncolytic herpes simplex virus HF10 (canerpaturev) promotes accumulation of CD8+ PD-1- enriched tumor microenvironment. Int J Cancer. 2021; 1 (149):
214-227.
101. Boorjian SA, Alemozaffar M, Konety BR, et al.
Intravesical nadofaragene firadenovec
gene therapy for BCG-unresponsive non-muscle-non-invasive bladder cancer: A
single-arm, open-label, repeat-dose clinical trial. Lancet. 2021; 1: 107-117.
102. Araki H, Tazawa H, Kanaya N, et al. Oncolytic
virus-mediated p53 overexpression promotes immunogenic cell death and efficacy
of PD-1 blockade in pancreatic cancer. Mol Ther Oncolytics.
2022; 27: 3–13.
103. Huang FY, Wang JY, Dai SZ, et al. A recombinant oncolytic
Newcastle virus expressing MIP-3a promotes systemic antitumor immunity. J Immunother Cancer. 2020; 8 (2): e000330.
104. Hinterberger M, Giessel R, Fiore G, et al. Intratumoral virotherapy with 4-1BBL armed modified
vaccinia Ankara eradicates solid tumors and promotes protective immune memory.
J Immunother Cancer. 2021; 9 (2): e001586.
105. Rahman MM, McFadden G. Oncolytic viruses: newest frontier
for cancer immunotherapy. Cancers. 2021; 13 (21): 5452.
106. Liu, J, Fu, M, Wang, M. et al. Cancer vaccines as promising
immuno-therapeutics: platforms and current progress. J Hematol Oncol. 2022;
15 (1): 28.
107. Sobhani, N, Scaggiante, B,
Morris, R, et al. Therapeutic cancer vaccines: From biological mechanisms and
engineering to ongoing clinical trials. Cancer Treat Rev. 2022; 109: 102429.
108. Li, L. et al. Optimized polyepitope neoantigen DNA
vaccines elicit neoantigen specific immune responses in preclinical models and
in clinical translation. Genome Med. 2021; 13 (1): 56.
109. Bauer J, Kohler N, Maringer Y, et al. the oncogenic
fusion protein DNAJB1-PRKACA can be specifically targeted by peptide-based
immunotherapy in fibrolamellar hepatocellular carcinoma. Nat Commun. 2022; 13: 6401.
110. Fritah H, Rovelli R,
Chiang CL, Kandalaft LE. The current clinical landscape of personalized cancer
vaccines. Cancer Treat Rev. 2022; 106: 102383.
111. Krause, W. mRNA—From COVID-19 Treatment to Cancer
Immunotherapy. Biomedicines. 2023; 11 (2): 308.
112. Linch M, Papai Z,
Takacs I, et al. 421 A first-in-human (FIH) phase I/IIa
clinical trial assessing a ribonucleic acid lipoplex (RNA-LPX) encoding shared
tumor antigens for immunotherapy of prostate cancer; preliminary analysis of
PRO-MERIT. J. Immunother. Cancer. 2021; 9 (2): 451.
113.
Jansen Y, Kruse
V, Corthals J, et al. A randomized controlled phase
II clinical trial on mRNA electroporated autologous monocyte-derived dendritic
cells (TriMixDC-MEL) as adjuvant treatment for stage
III/IV melanoma patients who are disease-free following the resection of macrometastases. Cancer Immunol. Immunother.
2020; 69: 2589–2598.
114. Beck JD, Reidenbach D, Salomon N, et al. mRNA
therapeutics in cancer immunotherapy. Mol. Cancer. 2021; 20 (1): 69.
115. Bowen WS, Svrivastava AK, Batra
L, Barsoumian H, Shirwan H. Current challenges for
cancer vaccine adjuvant development. Expert Rev Vaccines. 2018; 17 (3): 207–215.