A promising
therapeutic approach of dendritic cell vaccines for ovarian cancer
Asal Abolghasemi Fard 1,
Alireza Emamvirdizadeh 2 *
1 Department of Cellular and Molecular
Biology, Faculty of Modern Sciences and Technologies, Tehran Medical Sciences,
Islamic Azad University, Tehran, Iran
2 Department of Genetics, Faculty of
Modern Sciences and Technologies, Tehran Medical Sciences, Islamic Azad
University, Tehran, Iran
Corresponding Authors: Alireza Emamvirdizadeh
* Email: a.emamvirdizadeh@iau-tnb.ac.ir
Abstract
Ovarian cancer (OC) remains one of the most lethal gynecological
malignancies, primarily due to its often late-stage diagnosis and the
development of resistance to conventional therapies. In recent years,
significant advancements in immunotherapy have highlighted the potential of
dendritic cell (DC) vaccines as a novel therapeutic approach. This review aims
to thoroughly evaluate the current landscape and the future potential of DC
vaccinations for OC therapy. Recent Studies have provided evidence that DC vaccines
can generate specific T-cell responses, thereby enhancing the immunogenicity of
ovarian tumors. Furthermore, combining DC vaccines with other therapeutic
modalities, such as checkpoint inhibitors and chemotherapy, has shown
considerable promise in overcoming the immune evasion mechanisms employed by
tumors. However, several challenges remain, including optimizing antigen
selection, improving DC maturation and migration, and countering tumor-induced
immunosuppression. Continued research is essential for fully unlocking the
potential of DC vaccines in improving outcomes for ovarian cancer patients.
Keywords: Ovarian Carcinoma, DC subsets, Immunotherapy, Dendritic Cell Vaccine,
Hereditary Ovarian Cancer
Graphical
abstract
Introduction
Ovarian
Cancer (OC), a malignant tumor that develops in the ovaries, is often referred
to as the "silent killer" due to its subtle symptoms and late
diagnosis. It ranks as the seventh leading cause of cancer-related deaths in
women and is the deadliest among gynecologic cancers (1, 2). Among female
patients, ovarian cancer makes up 4% of all malignancies and 25% of cancers
affecting the female reproductive system. It leads to 5% of female deaths and
more than 50% of deaths caused by cancer of the female genital tract. The main
types of ovarian carcinomas are serous (40%), mucinous (10%), endometrioid
carcinoma (20%), undifferentiated carcinoma (10%), and clear cell tumors (3).
Several
elements contribute to the prognosis of a tumor, including tumor margin,
vascular invasion, tumor grade and stage, expression of oncogenes, and the
presence of estrogen and progesterone receptors (3, 4). Immune cells within the
tumor, such as Dendritic Cell (DCs), may also serve as a prognostic factor. DCs
are a rare immune cell population found in tumors and lymphoid organs, but they
play a central role in initiating antigen-specific immunity and tolerance.
Manipulating DCs has the potential to effectively induce anti-tumor immunity
(5). DCs play a crucial role in the immune system by enhancing immunity or
inducing tolerance. This is achieved through the presentation of antigens to T
cells, and the delivery of immunomodulatory signals via direct cell-to-cell
interactions and the secretion of cytokines (6).
The functions of DCs are influenced by their capacity to sense and
respond to environmental stimuli, which are detected through various receptors
located on the cell surface and within the cell for cytokines,
pathogen-associated molecular patterns (PAMPs), and damage-associated molecular
patterns (DAMPs). Recent research underscores the unique functions of DC
subsets in antitumor immune responses, offering important insights for therapy
and making them a promising tool in vaccine development, especially for
diseases like cancer, infectious diseases, and autoimmune disorders (7, 8). To
initiate and maintain protective anti-tumor immunity, optimal DC function is
necessary. However, aggressive cancers can effectively evade immune control by
impairing normal DC functions (9). The understanding of DC subsets and their
functions has predominantly been shaped by research in mice; however, there is
an increasing interest in exploring the biology of human DCs (10,11). This
article will delve into the primary functions of DCs in cancer immunology and
examine the potential therapeutic strategies involving the targeting of DCs in
vaccines for patients with OC. Despite all these therapeutic advances,
approximately 80–85% of the advanced-stage patients still relapse, indicating
the urgent need for novel therapies against OC.
1. Ovarian
Carcinoma
Among
women, OC ranks seventh in terms of global cancer diagnosis, following breast,
colorectal, lung, endometrial, thyroid, and non-Hodgkin's lymphoma (12).
Approximately 239,000 new cases and 152,000 deaths are reported annually (13).
Eastern and Central Europe record the highest rates, with 11.4 per 100,000 and
6.0 per 100,000, respectively (6, 13). As a worldwide concern, late diagnosis
and the absence of an effective screening strategy contribute to the complexity
of the issue. Moreover, newly diagnosed cancer is commonly managed through
cytoreductive surgery and platinum-based chemotherapy (14).
Three
main cell types - epithelial cells, stromal cells, and germ cells - are
responsible for the formation of ovarian tumors, whether they are benign or
malignant. In developed nations, more than 90% of malignant tumors are
classified as sex cord-stromal tumors. While most epidemiologic research,
including this review, emphasizes epithelial OC (15). For instance, granulosa
ovarian tumors are derived from epithelial cells. Around 5% to 6% of tumors are
cell tumors, like thecomas, whereas germ cell tumors, such as teratomas and
dysgerminomas, make up approximately 2% to 3% (13, 16). OC is classified into
five distinct histological subtypes, each with identifiable risk factors, cells
of origin, molecular compositions, clinical features, and treatments. These
subtypes include high-grade serous (HGSOC; 70%), endometrioid (ENOC; 10%),
clear cell (CCOC; 10%), mucinous (MOC; 3%), and low-grade serous (LGSOC;
<5%) (15) (Figure 1).
Among
these subtypes, high-grade serous carcinoma is the most commonly diagnosed. In
contrast, HGSC shares similarities with high-grade endometrioid carcinoma.
Among the less frequent histologies, small-cell
carcinoma is distinguished by its highly aggressive behavior, often seen in
younger women who are diagnosed around the age of 25. The tissue origin of this
type of cancer remains uncertain. Additionally, carcinosarcoma, another type of
aggressive cancer, is also recognized in certain cases (14, 17). The exact
cellular origin and pathogenesis of OC are still unclear. It is interesting to
note that a significant proportion of tumors seem to arise from different
gynecological tissues, primarily affecting the ovary. Studies on morphology and
genetics have shown that the fallopian tube epithelium is the origin of both
high- and low-grade serous neoplasms. Furthermore, endometriotic cysts are
connected to CCOC and ENOC, while MOC is thought to come from transitional cell
nests at the tubal-mesothelial junction. HGSOC and LGSOC are believed to stem
from the tubal epithelium, albeit through separate pathways (18).
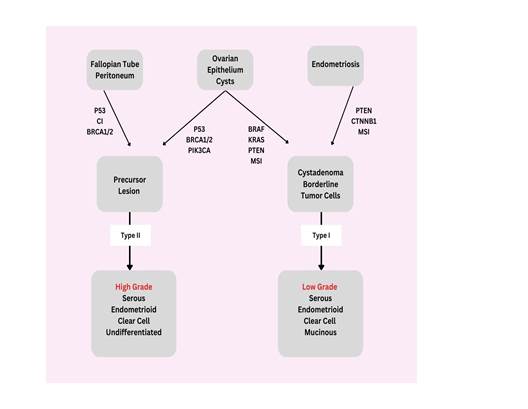
Figure
1.
Two-pathway concept of ovarian cancer development (1).
The
presence of serous tubal intraepithelial carcinomas, or tubal lesions in the
fimbriated end of the fallopian tube, show similarities in morphology and TP53
signatures to tumors. This suggests that the progression of cancer may begin at
these tube lesions and advance rapidly into the ovary (2-4,18). LGSOC tumors
are identified across a range that signifies a clear progression from benign
serous cystadenoma to borderline serous tumors and finally low-grade carcinoma.
The glands of epithelial inclusion, believed to have derived from the
cystadenoma, are situated in the ovary but display traits similar to those of
the fallopian tube, indicating they may have developed from transplanted tubal
epithelium (5,16,18). Current epidemiological studies on OC are delving deeper
into the investigation of etiologic factors based on histopathologic and
molecular subtypes, utilizing the approach of "molecular pathological
epidemiology." The evidence from these studies shows that several risk
factors have distinct correlations with the primary histotypes
(7, 18).
2. Hereditary
and Genetic of Ovarian Cancer
Hereditary
OC syndromes appear to be genotypically and phenotypically heterogeneous
diseases characterized by variable clinical courses (18,19,20). The role of
genetic factors in the pathogenesis of OC is well documented. Hereditary OC
accounts for at least 5–15% of ovarian carcinomas (18,19). OC risk is
influenced by a range of distinct hereditary genetic anomalies (3,21); for
example, mutations in the BRCA1 and BRCA2 genes, which are linked to breast
cancer, contribute to approximately 90% of OC cases in individuals with a
family history of hereditary breast-ovarian cancer. Individuals with BRCA1
mutations have a lifetime risk of OC of approximately 40–50%, while those with
BRCA2 mutations have a risk of 20–30% (21). Furthermore, alterations in the
BRCA genes elevate the susceptibility to various types of cancer, which include
breast cancer, specifically BRCA1 and BRCA2 mutations; pancreatic cancer linked
to BRCA2 mutations; prostate cancer associated with BRCA2 mutations; melanoma
also connected to BRCA2 mutations; and potentially serous
endometrial cancer related to BRCA1 mutations (7,21). Studies have shown that
the presence of deleterious mutations in BRCA1/2 and other genes involved in
repairing double-strand DNA breaks is significantly correlated with an
increased susceptibility to HGSOC, although these mutations can manifest in
other subtypes of tumors as well (21,
22).
Apart from BRCA1 and BRCA2, there are other genetic mutations in
genes involved in DNA repair that can raise the chances of developing OC,
including genes within the Fanconi anemia-BRCA pathway like RAD51C, RAD51D,
BRIP1, BARD1, and PALB2 (22,23). The presence of inherited mutations in other
genes involved in DNA repair, namely CHEK2, MRE11A, RAD50, ATM, and TP53, may
also contribute to an increased likelihood of OC development
(7, 22, 23).
Other
inherited disorders, such as Lynch syndrome, are also responsible for an
additional 10–15% of hereditary ovarian carcinomas (18,20). The syndrome is
characterized by the inheritance of a germline mutation predominantly caused by
mutations in four mismatch repair genes (MLH1, MSH2, MSH6, and PMS2),
representing 65–85% of cases (23,24). Studies have provided evidence that
individuals with Lynch syndrome are more likely to develop endometrioid and
clear-cell carcinomas in comparison to the expected occurrence in cases of
sporadic OC (7, 25). Despite the involvement of both the BRCA and DNA mismatch
repair pathways in DNA repair, the specific reasons behind the occurrence of
cancers in particular organs associated with these inherited mutated genes
remain understudied (26).
3. Dendritic cells
Subsets and Functions in OC
The
prognosis of OC is dependent on a variety of factors, including tumor margin,
vascular invasion, tumor grade and stage, oncogene expression, and estrogen and
progesterone receptor status (9,26). Additionally, the presence of immune cells
within the tumor, such as DCs, can serve as an additional prognostic factor
(10,27). Considered the most effective antigen-presenting cells, DCs serve as a
bridge between the immune system of the host and tumor cells, reflecting their
intricate interaction (11,12,27), and despite their limited presence in the
body, these cells play a crucial role in triggering antigen-specific immunity
and tolerance, making them the predominant cell type (8).
DCs are developed from CD34+ hematopoietic stem cells situated in
the bone marrow. Following this, they undergo differentiation into diverse
subtypes in the peripheral blood and nonlymphoid organs and tissues, ultimately
reaching maturation in the lymphoid organs (13-15). Immature dendritic cells
show lower levels of toll-like receptors (TLRs), major histocompatibility
complex (MHC) molecules, costimulatory molecules, and adhesion molecules.
Consequently, these cells are found in peripheral tissues and have restricted
antigen-presenting functions (7, 9, 21).
TLRs
are recognized as the key receptors involved in the detection of PAMPs and
DAMPs (15,28). Through the activation of DCs, PAMPs stimulate the innate immune
response, which serves as a crucial defense against infectious diseases. In the
context of tumors, DCs are activated in response to DAMPs released by tumor
cells via TLR signaling (12,16,26). Immature DCs respond to chemokine ligands
CCL19 and CCL21 by migrating towards the lymph nodes. The maturation of these
DCs involves the up-regulation of chemokine receptors CCR7 and CCR8, which
enhance their migration (17). While situated in the lymph nodes, they undergo a
progressive change into a mature state, marked by an elevated expression of MHC
I molecules, MHC II molecules, costimulatory molecules, and adhesion molecules
(17,18,28). There are three main subsets into which DCs can be divided:
conventional or classical DCs (cDCs, also called
myeloid DCs), monocyte-derived DCs (moDCs), and
plasmacytoid DCs (pDCs) (8,12,14). cDCs can be further classified as cDC1, cDC2, and migratory
DCs (migDCs) (12,13,29) (Figure 2).
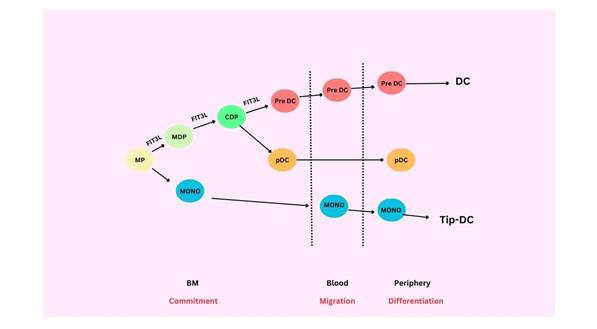
Figure
2.
Dendritic cell and monocyte origin and development (29).
3.1
cDC1
cDC1
serves as the primary DC subtype responsible for regulating cancer
immunotherapy responses by activating CD8+ T cells via the antigen
cross-presentation mechanism (9, 13). They are of utmost importance in
facilitating the early activation of CD4+ T cells against tumor-derived
antigens via MHC-II, and their role in delivering CD4+ T cell assistance to
CD8+ T cells cannot be underestimated (18.29). However, the absence of CDC1s
during viral infections disrupts the proper differentiation of memory CD8+ T cells,
resulting in unfavorable outcomes (12, 19). cDC1s are also potent in their
production of interleukin-12 (IL-12) and have the capability to induce NK and
CD8+ T-cell cytotoxicity as well as the generation of interferon-gamma (IFNγ) (19). IFNγ contributes to a
positive feedback loop that increases cDC1-mediated IL-12 production,
ultimately bolstering antigen cross-presentation
(20).
3.2. cDC2
Classically,
cDC2 releases IL-10, IL-12, IL-23, and TNF-b to promote the development of CD4+
helper T cells (9, 13), particularly T helper type 2 (Th2) (18,28) and T helper
17 (Th17) cells (20, 21). These cells are distinct from cDC1s and are unable to
functionally fill in for cDC1 deficiencies (12). Studies have indicated that
cDC2s can increase the activation of existing CD8+ T cells during anti-CD40
therapy (22). The understanding of cDC2 functions is obstructed by three
fundamental hindrances. First, the absence of a definitive marker specific to
cDC2 poses a challenge in elucidating the contribution of cDC2s to tissue
immune responses in vivo through conditional depletion models. Second, a
resemblance can be seen in the present cDC2 markers and phenotypic
characteristics with alternative myeloid compartments such as moDCs and macrophages, which poses challenges in isolating
the specific contribution of cDC2s in functional inferences compared to other
myeloid cells (23,30). Third, the cDC2 compartment is known for its
heterogeneity, housing diverse sub-populations. This suggests that each subset
within this compartment may possess unique functionalities (10, 24, 25).
Various immune contexts have led to the identification and categorization of
cDC2 sub-populations, with some overlap in their characteristics. To gain a
better understanding, further investigation is necessary. This is especially
important for DC vaccines, as targeting the most potent cDC2 subpopulation
could potentially improve patient outcomes compared to targeting the entire
cDC2 compartment, which may contain some anti-inflammatory sub-populations (23,
29, 30).
3.3.
migDC
Migratory
DCs, also known as migDCs, DC3, mregDC,
or LAMP3+ DCs, are a unique type of fully developed cells that can be found in
both cDC1s and cDC2s when they detect or absorb antigens (25, 26). MigDCs are dendritic cells found in non-lymphoid tissues
that travel to the tdLN through the lymphatic system
instead of the bloodstream. In inflammation, migDCs
loaded with antigen move to T-cell regions in LNs to activate CD4+ and CD8+ T
cells. They upregulate MHC-II and costimulatory molecules and secrete inflammatory
cytokines to enhance T-cell responses (27, 28).
4. Dendritic
Cell Dysfunction in The Tumor Microenvironment
Within
OC lesions, there is a notable presence of DC infiltration; nevertheless, the
infiltrated DCs exhibit a decreased efficacy in antigen presentation owing to
DC tolerance. This tolerance is distinguished by the reduced expression of
costimulatory molecules on the DC cell surface, leading to a compromised
antigen-presenting capability. DCs can assist tumor cells under specific
conditions (27, 30). In the absence of tumors, hematopoietic precursors
differentiate into progenitors that further specialize into immature DCs.
Immature DCs mature and specialize in antigen presentation after meeting an
antigen or "danger signal." Nonetheless, differentiation of DCs is
commonly disrupted in the tumor microenvironment, resulting in a buildup of
defective and immature DCs. In mouse melanoma, tumor-infiltrating DCs contained
both myeloid and plasmacytoid DC populations (31). Most of these DCs appeared
immature, but about a third expressed a mature phenotype (32).
DC
dysfunction can be impacted by immune checkpoint signaling. When PD-1 on T
cells interacts with PD-L1 on tumor cells, it can lead to the death of T cells.
PD-1 inhibitors could enhance the antitumor effect of DCs in OC (33). Through
the release of TGF-b and PGE2 into the microenvironment, OC cells can stimulate
the upregulation of PD-L1 in DCs, which strengthens their ability to suppress
the immune response of T cells (33,34). Immunosuppressive cells and specific
DCs have a direct interaction that affects the body's ability to combat tumors.
In ovarian carcinoma, the interaction between pDCs
and regulatory T cells (Treg cells) is facilitated by the expression of the
ICOS ligand, leading to tumor progression (34). Additionally, insulin-like
growth factor (IGF) influences dendritic cells (DCs) in ovarian cancer,
impacting cell proliferation, protein synthesis, and growth through the
activation of the RAS-ERK and PI3K-AKT pathways. In the presence of IGF, DCs
fail to mature and secrete higher levels of IL-10 and TNF-a, considered
immunosuppressive factors in the OC microenvironment (35, 36). The insulin-like
growth factor type I receptor (IGF1R) is prominent in OC. This receptor has a
negative correlation with the differentiation of DCs into cDCs.
By utilizing IGF1R inhibitors, the DC-mediated antitumor effect can be rebuilt.
This suggests that the IGF axis may be responsible for inducing dysfunction in
DCs (36,37). To conclude, immunosuppressive signals contribute to DC
dysfunction in OC. By infusing functional DCs into the body, they can engage
with T cells in lymph nodes rather than the tumor microenvironment, potentially
restoring their ability to present tumor antigens and induce antitumor effects (38,
39).
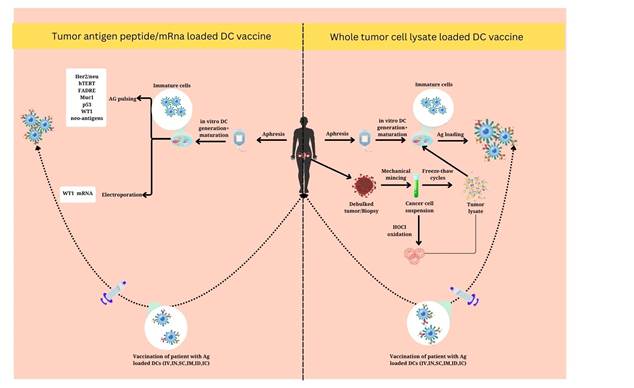
5. DCs Vaccine in OC
Cancer
vaccines are divided into various groups based on how they deliver the chosen
TAAs. These groups include cell-based vaccines, peptide/protein vaccines, and
genetic vaccines (Table 1) (31, 35).
5.1 cell-based
vaccines
Cell-based
vaccines can use DCs to help connect innate and adaptive immunity (40). The
goal is to trigger cytotoxic T lymphocytes to target and destroy cancer cells
using tumor antigens (41,42). DCs are essential for immunosurveillance, which
underscores the immune system's vital role in recognizing and removing
pathogens and cancer cells. However, the slow progression of malignancy during
its initial phases can result in occasional failures of immunosurveillance
(39). In the early stages, tumors can occasionally inhibit an immune response
or fail to produce the essential signals for immune system activation.
Cell-based vaccination aims to fix this problem by reversing the immune
system's lack of knowledge about cancer cells (43).
Adjuvant DC vaccines have proven to be effective in the long run
for people with melanoma, glioblastoma, prostate cancer, and renal cell
carcinoma. However, it is important to note that these improvements have only
been demonstrated in a small number of patients (44, 45). DC vaccination is
considered safe and typically causes fewer side effects than chemotherapy and
ICBs (39). Choosing the appropriate DC subtypes is a key factor in successful
vaccination. The chosen subtypes of autologous DC used in vaccine production
display different levels of antigen-presenting potential, potentially
influencing the effectiveness of DC vaccines. In the study of DC vaccines for
tumors, scientists select particular DC subtypes from peripheral blood cells
using apheresis. These subtypes, such as MoDCs, cDCs, and Langerhans cell-type DCs, are assessed in
preclinical and clinical studies (36, 45). Various DC subtypes are being
targeted to improve immune responses against tumors in vaccines that target DC
within and outside the body, and these may vary depending on the cancer types
(46,47). To manufacture vaccines that target DCs in the body, there is no need
for apheresis to gather autologous DCs. Instead, specific antigens that target
receptors on DCs are injected directly into the body. For example, the vaccine
CDX-1401 is formulated to target DEC205+ cDC1s in multiple tumors, such as OC.
This vaccine includes the DEC205 antibody fused with NY-ESO-1 and a TLR agonist
(48, 49). The development of vaccines that target DCs externally involves the
use of peripheral blood cells obtained through apheresis
(50).
MoDCs are the
preferred subtype for this purpose due to the limited number of DCs in
peripheral blood cells for vaccine production. On the other hand, a larger
number of DCs can be generated from monocytes when cultured in vitro compared
to other sources (45). When it comes to vaccinations, cDCs
are more potent than MoDCs in inducing long-lasting
and broad immune responses. Furthermore, cDCs can
enhance the efficacy of immune checkpoint inhibitors (51). The presence of
cDC1, cDC2, and pDC in OC has been previously noted.
The ratio of cDC and pDC
varies in peripheral blood, ascites, and tumor sites. Among DC subsets, pDC is most frequently found in ascites (40) and tumor
sites (10), while cDC is more abundant than pDC in the peripheral blood (35). This indicates that
peripheral blood could be a valuable resource for the production of DCs (52).
Due to the limited number of cDCs available for
vaccine manufacturing, MoDCs are commonly used in
clinical studies on DC vaccines (27). After isolation from peripheral blood
using apheresis, mononuclear cells are cultured in vitro with GM-CSF and IL-4
for a specific duration. The evaluation of markers on DCs, including CD11c+,
HLA-DR+, HLA-ABC+, CD40+, CD80+, CD83+, CD86+, and CCR7+, is performed to
monitor the cellular composition of the DC vaccine (53). However, these markers
are not effective in distinguishing MoDCs from other
DC subtypes, resulting in the DC vaccine being a combination of DCs and a small
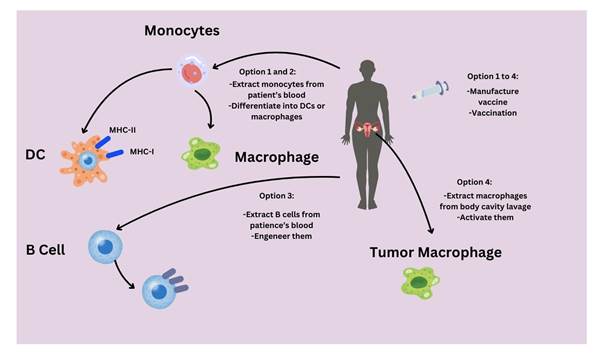
proportion of other peripheral blood cells (11, 27) (Figure 3).
5.2
Peptide/Protein-Based Vaccines
Autologous
cancer vaccines, such as DCs or whole tumor cells, are limited by the need for
patient samples and the complex process of making personalized vaccines.
Recombinant vaccines have an advantage in this respect. Peptide- or
protein-based vaccines typically utilize specific TAAs and are given with an
adjuvant or immune modulator to enhance uptake by DCs (3,53). Many different
peptides have been experimented within OC to find out if they can target
HER-2/neu. HER-2/neu is a member of the HER/EGFR/ERBB family, and if it's
amplified in breast cancer, it makes the cancer more aggressive. That's why
it's an important target for around 20%–30% of patients (54). The presence of
HER-2/neu overexpression or amplification has been detected in OC cases (19),
suggesting it as a potential target for cancer vaccination. Nevertheless,
studies using HER-2/neu peptides have not shown any immune response (14, 36),
and there is no clinical data available (31). The most efficient outcomes in OC
treatment through peptide-based vaccines have been achieved by employing a
personalized peptide vaccine (PPV). This method consists of mixing four
peptides (selected from a set of 31) that have been tested for immune response
in every patient and then injecting them subcutaneously in Montanide
ISA51VG (19, 31, 35). The study revealed that platinum-sensitive patients had a
median survival time of 39.2 months, while platinum-resistant patients had 16.2
months. Standard of care patients had 18–30 months (platinum-sensitive) and
8–12 months (platinum-resistant). Notably, PPV not only enhanced immune
responses to specific peptides but also extended to other peptides, resulting
in longer survival (50). The findings indicated that selecting and
administering vaccine antigens based on the patient's pre-existing immunity
before vaccination could extend overall survival in advanced OC patients (55).
Figure
3. An
overview of dendritic cell vaccination strategies used in ovarian carcinoma.
Ag, antigen; HOCl, hypochlorous acid; IV,
intravenous; IN, intranodal; SC, subcutaneous; ID, intradermal; IC,
intracutaneous.
5.3
Genetic Vaccine
The
use of genetic vaccines, whether they are DNA, RNA, or virus-based, can
activate the expression of chosen TAAs within somatic cells like keratinocytes,
myocytes, or DCs that infiltrate muscle or skin at the vaccination site. This
can result in either cross-priming or direct antigen presentation to
infiltrating T-cells. Genetic vaccines make it easy to deliver multiple
antigens in one immunization, activate different branches of immunity, and have
a more cost-effective and standardized manufacturing process (30). Two viral
vaccines have been tested for OC: One team is concentrating on the
"cancer-testis" antigen NY-ESO-1, which has been integrated into
vaccinia (rV) as the initial vaccine and fowlpox (rF) as the follow-up
vaccine. The second genetic vaccine tested for ovarian cancer, PANVAC-C +
PANVAC-V, is a Poxviral vaccine. It involves engineering CEA-MUC1-TRICOM (B7.1,
ICAM-1, LFA-3) into vaccinia (PANVAC-V) as the prime and fowlpox
(PANVAC-C) as the booster vaccination (37, 38).
A Phase I clinical trial with 25 patients with CEA- or MUC1-expressing
metastatic cancers, including three with OC, showed limited clinical activity.
Ongoing studies are investigating different genetic vaccines for treatment
(56,57,58).
Table
1.
Published results from therapeutic vaccines tested in ovarian cancer from 2000
to 2024.
|
Class |
Name |
Description |
Clinical Development Phase |
No. of Pts (OvCa Pts) |
Clinical Result |
Ref |
|
DCs |
APCEDEN |
DCs loaded with whole-tumor lysate |
Phase II |
38 pts (9 OvCa pts) |
No CR observed; ORR was 28.9% (11/38) and irRC
was 42.1% |
(35) |
|
DCVax-L |
DCs
loaded with autologous oxidized tumor lysate, combined with bevacizumab and
metronomic Cy |
Pilot |
6 OvCa pts |
4/6
pts (66%) achieved clinical benefit (including 2 PR and 2 SD) |
(37) |
|
|
OCDC |
DCs loaded with autologous oxidized tumor lysate |
Pilot |
5 OvCa pts |
2/5 pts (40%) demonstrated PFS2 > PFS1 |
(30) |
|
|
DC-MFP |
DCs
loaded with |
Phase
I |
9
pts |
2/9
pts (22%) in |
(33) |
|
|
DC-wtl |
DCs loaded with crude |
Phase I |
8 pts |
Data suggested |
(33) |
|
|
Lapuleucel-T, |
DCs
loaded with BA7072, |
Phase
I; |
18
pts |
2/18
pts (11%) had SD |
(24) |
|
|
HER-2/neu; MUC1 |
DCs loaded with synthetic |
Phase I; |
10 pts |
No data |
(24) |
|
|
hTERT; |
DCs
loaded with synthetic |
Phase
I/II |
14
OvCa pts, |
3
years-OS was 90%; |
(24) |
|
|
WT-1; MUC1; |
DCs loaded with syntheticpeptides
derived from |
Phase II |
56 OvCa pts |
DCR and ORR were 29% |
(35) |
|
|
Peptides/ |
Mixture
of |
Predesigned
peptides vs. |
Pilot |
14
pts |
No
clinical response |
(41) |
|
Mixture of |
OvCa-associated
peptides |
Phase I |
9 OvCa pts, |
One participant |
(41) |
|
|
Mixture |
OvCa-associated
peptides |
Pilot |
15
pts |
With
median follow-up |
(42) |
|
|
HER-2/neu |
Epitope p369–377, |
Phase I; |
6 pts |
No data |
(42) |
|
|
HER-2/neu-ICD |
ICD
protein, aas 676–1255, |
Phase
I; |
29
pts |
No
data |
(45) |
|
|
NY-ESO-1 |
Epitope p157–170, |
Phase I |
18 OvCa pts, |
Median PFS of 19.0 mo |
(46) |
|
|
NY-ESO-1
OLP |
NY-ESO-1
overlapping |
Phase
I |
28
OvCa pts |
Pts
NY-ESO-1+ receiving |
(57) |
|
|
NY-ESO-1 protein |
NY-ESO-1 protein + |
Phase I |
12 OvCa pts |
5/10 (50%) pts had SD |
(57) |
|
|
P53 |
Wt p53: 264–272
peptide |
Phase
II; |
21
OvCa pts, |
No
significant difference |
(68) |
|
|
P53-SLP |
Ten synthetic peptides |
Phase II |
18 OvCa pts |
2/18 (11%) of pts with |
(35) |
|
Flt3-L |
Truncated
glycoprotein |
Pilot |
15
pts |
No
objective responses |
(35) |
|
|
PPV |
Personalized peptide |
Phase II |
42 OvCa pts |
Median survival time |
(58) |
|
Whole |
Fang
vaccine, |
Autologous
tumor cells |
Phase
I |
27
pts |
23/26
pts (88%) showed |
(32) |
|
Genetic |
PANVAC-C + |
|
Pilot; |
25 pts |
1 OvCa pt (1/25: 4%) |
(39) |
|
rV-NY-ESO-1
+ |
NY-ESO-1
engineered into |
Phase
I; |
36
pts |
7/9
pts with stage |
(39) |
Abbreviations:
aas, aminoacids; CR,
complete response; DCR, disease control rate (SD + PR + CR); irRC, immune-related response criteria; mo,
months; MST, median survival time; ORR, objective response rate (PR + CR); OS,
overall survival; PD, progressive disease; PFS, progression free survival; PR,
partial response; Pt(s), patient(s); SD, stable disease; TTP, time to
progression.
6.
DCs in the cancer therapy
DCs
have the potential to influence the efficacy of cancer therapies currently
employed in clinical practice. This review delves into the impact that DCs can
have on the response to such treatments (7).
6.1
Chemotherapy and DCs
Traditionally,
chemotherapeutic treatments such asbortezomib,
doxorubicin, epirubicin, idarubicin, and Mitoxantrone
and oxaliplatin have long been thought to provide anti-cancer benefits by
either directly killing cancer cells or causing a permanent cessation of the
cell cycle, and these responses depend on DCs (16,59). It was believed that
chemotherapy could target rapidly dividing cells, including immune cells, and
cause immunosuppression. Many chemotherapy drugs used in clinics are not
immunogenic or have immunosuppressive side effects. They can directly inhibit
or kill effector cells or indirectly cause energy or immune paralysis. As a
result, the immune system's role in anticancer therapy has been largely ignored
(18). It is now commonly believed that certain chemotherapy drugs and
anticancer medications can trigger the body's immune system to fight tumors (19,
60).
One
way they do this is by making tumor cells more visible to the immune system,
which leads to an immune response against the tumor. This has been shown in
experiments with mice that have a healthy immune system. Additionally,
immunogenic cell death (ICD) may be induced by specific physical methods like
UV-C irradiation, hypericin-based photodynamic therapy, and high hydrostatic
pressure, while certain oncolytic viruses possess the intrinsic capacity to
initiate ICD. These were among the chemotherapeutic drugs used in clinical
practice: anthracyclines (doxorubicin, epirubicin,
and idarubicin), mitoxantrone, oxaliplatin, CTX, and bleomycin (BLM) (29, 60).
The efficacy of these stimulants in triggering an immune response against
tumors relies on the development of adaptive stress reactions that facilitate
the synchronized release of endogenous danger signals from apoptotic cells.
These signaling molecules, referred to as DAMPs, interact with various
receptors found on dendritic cells to activate the adaptive branch of the
immune system (61). Multiple DAMPs have been identified as characteristic
elements of ICD, specifically the initial presentation of the endoplasmic
reticulum (ER) chaperone calreticulin (CRT) and heat-shock proteins (HSPs)
HSP70 and HSP90; the spontaneous release of molecules like high mobility group
box 1 (HMGB1); and the excretion of adenosine triphosphate (ATP) (10, 31, 62).
In addition, some chemotherapy drugs can induce tumor cells to produce type I
interferons (IFNs). Although type I IFNs are not DAMPs specifically, they have
strong immune-boosting effects and are crucial for chemotherapy-induced cell
death to be recognized as immunogenic. To conclude, the activation of the
immune system is supported by DAMPs, as demonstrated in many in vitro tumor
cell line models and in vivo mouse immunization experiments. Recent reports
also suggest that monitoring DAMPs in cancer patients may have prognostic or
predictive value (30, 32).
6.2
Radiation therapy and DCs
Highly
proliferating cells are the preferred targets of radiation treatment. While
this therapy's primary function is to directly kill cancer cells, this
explanation falls short of explaining the therapy's overall effect on tumor
growth. Radiation therapy's anti-tumor efficacy also involves local bystander
effects, such as the release of DAMPs and cytotoxic mediators, the alteration
of the immunological TME, and the in situ generation
of reactive oxygen species (63, 64). Additionally, radiation therapy can generate
distant effects, referred to as out-of-field or abscopal effects, that are
correlated with the promotion of systemic immune responses against cancer,
facilitated by the induction of immunogenic cell death and the activation of
CD8+ T cells by cDC1 Following radiation therapy, cancer cells release
cytosolic DNA that acts as a DAMP, signaling through cGAS-STING
to stimulate type I interferon production by DCs, thus aiding in antitumor
immunity (55). However, high radiation doses prompt the expression of DNase
TREX1, which breaks down cytosolic DNA, limiting interferon production and the
immunostimulatory impact on cDC1s (54).
6.3
Small-molecule inhibitors and DCs
Small-molecule
inhibitors have been developed to target important oncogenic signaling pathways
such as STAT3 and mitogen-activated protein β-catenin signaling (26). These
pathways are associated with decreased cDC1 tumor infiltration and a lack of
response to immune checkpoint blockade therapy. Nevertheless, the transfer of
preactivated in vitro-generated cDC1-like cells with poly(I:C)5 was effective
in reversing this non-responsiveness (8). Moreover, the combination of
vaccination with naturally existing cDC1s loaded with immunogenic cell
death-derived whole tumor antigen and anti-PD1 treatment reveals a synergistic
outcome. The synergy between TLR-induced activation of DCs and ICB can be
heightened by FLT3L-induced expansion of DC populations. Recent discoveries
suggest that cDC1 is vital for cross-priming, as evidenced by WDFY4-deficient
mice being incapable of rejecting immunogenic tumors due to a defect in a
vesicular transport pathway necessary for cross-presentation (18, 32).
Enhancing the function of DCs may result in improved and expanded
responsiveness to ICB regimens. Both cGAS and STING
are crucial for intrinsic antitumor immunity and effective responses to
anti-PDL1, with DCs playing a key role in mediating these responses. (33). The
activation of type I interferons to stimulate cDC1s can potentially improve the
response to anti-PDL1 treatment, indicating a potential requirement for the
activation of tumor DCs to support effector T cell activity triggered by ICB.
Enhancing the production of chemokines like CXCL9 and CXCL10 by DCs, possibly
through epigenetic modifications, may also enhance the efficacy of ICB therapy
(32, 34).
7. Safety
of Dendritic Cell Vaccines
The
safety of DC vaccines has generated significant interest due to their potential
to modify immune cell, cytokine, and chemokine levels in the body. Thankfully,
the majority of OC patients involved in clinical studies have responded well to
DC vaccines. Most reported side effects are grade 1 or 2 and include common
symptoms like local skin reactions, fatigue, pain, flu-like symptoms, muscle
aches, fever, nausea, and vomiting (32). Numerous studies have reported serious
toxicity associated with DC vaccines, especially when used in combination with
other treatments. During the phase II trial of a p53 peptide cancer vaccine and
DC vaccine, every one of the 21 patients encountered a localized skin response.
Among the participants who were administered a combination DC vaccine
containing p53 peptide, a minimum of 3 patients documented lymphopenia and
fatigue (32). Additional toxicities related to the grade III/IV vaccine consist
of increased ALT and AST levels, fever, hypocalcemia, memory impairment, and
rigors (53). It is important to highlight that notable toxicity was connected
to the IL-2 treatment in the subgroup examination of this research. This was
noted during a phase I clinical trial of the DC vaccine for the maintenance
therapy of ovarian carcinoma (39, 40). Additionally, two patients suffered from
hypertension. More evidence is necessary to determine if these toxicities are
related to DC vaccines in OC patients undergoing chemotherapy. To conclude, DC
vaccines are usually well tolerated, but combining them with chemotherapy or
immunotherapy should be done carefully (Table 2) (23, 65,66).
Table
2.
Issues and challenges in cancer vaccine development (35).
|
Issues |
Challenges |
|
|
Personalised vaccination |
A. Development
of a robust and B. Generation of a strong immune
response against tumour antigens without inducing |
|
|
Immune
tolerance and |
A. Counteract mechanisms of immune evasion by cancer |
|
|
Immunotherapy
as single |
A. Development of rational
combination therapies |
|
|
Self-limited
immunity |
A. Maintenance of anti-tumour immune
response over time |
8. Future
of the DC Vaccines
DC
vaccines have exhibited promise in the realm of immunotherapy for ovarian
carcinoma. However, there exists untapped potential that necessitates
exploration through the use of new technologies, cohort studies, and
biomarkers. Tumor immunosuppressive signals have been found to impair dendritic
cells, leading to compromised immunological function and metabolism, thereby
resulting in issues related to antigen presentation and tumor growth (40). The
rise in popularity of personalized DC vaccines can be attributed to their
effectiveness in activating T cell responses that target tumor antigens
specific to individual patients, facilitated by next-generation sequencing and
bioinformatics analysis. Nonetheless, challenges such as complex preparation
techniques, limited tumor samples, and difficulties in selecting tumor antigens
need to be addressed. While clinical experiments have validated the safety of
DC vaccines, their efficacy varies depending on the manufacturing technique and
study strategy. The identification of an ideal biomarker is essential in this
context (41).
Conclusion
Advances
in cancer immunotherapy, notably for ovarian carcinoma, have demonstrated their
significance in the battle against cancer. Cykine
therapy, peptide vaccines, monoclonal antibodies, dendritic cell-based
vaccines, adoptive T cell transfer, immune checkpoint inhibitors, and various
nanoparticles are all being studied for ovarian cancer treatment. Combining
these tactics with individual therapy can help boost the immunological
response. However, there is still potential to enhance treatment options, such
as by studying tumor biology, immune-suppressive networks, and immunomodulatory
techniques. Polymeric and lipid-based nanoparticles are being created to
deliver antigens, immune stimulants, and immunoadjuvants in a sustained-release
manner. More research is needed to create accurate biomarkers and successful
treatment combinations.
Acknowledgments
The
author received no financial support for the research, authorship and/or
publication of this article.
Author
contribution
AAF wrote the main manuscript text and prepared the figures and tables.
AE and AAF reviewed and checked the manuscript
Conflict
of interest
The
authors declare that they have no competing interests.
Funding
The
author declares that there is no conflict of interest.
Consent
to Publication
We
the undersigned authors, give our consent for the publication of identifiable
details, which can include photograph and/or case history and/or details within
the text (“Material”) to be published in the above Journal and Article.
References
1. Huarte, E., et al., Depletion of
dendritic cells delays ovarian cancer progression by boosting antitumor
immunity. Cancer research, 2008. 68(18): p. 7684-7691.
2. Scarlett, U.K., et al., Ovarian cancer
progression is controlled by phenotypic changes in dendritic cells. Journal of
Experimental Medicine, 2012. 209(3): p. 495-506.
3. Martin-Lluesma,
S., et al., Are dendritic cells the most appropriate therapeutic vaccine for
patients with ovarian cancer? Current opinion in biotechnology, 2020. 65: p.
190-196.
4. Chae, C.-S., E. Teran-Cabanillas, and J.R.
Cubillos-Ruiz, Dendritic cell rehab: new strategies to unleash therapeutic
immunity in ovarian cancer. Cancer Immunology, Immunotherapy, 2017. 66: p.
969-977.
5. Aarnio, M., et al., Cancer risk in mutation
carriers of DNA‐mismatch‐repair genes. International journal of cancer, 1999.
81(2): p. 214-218.
6. Chen, W., et al., Cancer statistics in
China, 2015. CA: a cancer journal for clinicians, 2016. 66(2): p. 115-132.
7. Ricciardelli, C. and M.K. Oehler, Diverse
molecular pathways in ovarian cancer and their clinical significance. Maturitas, 2009. 62(3): p. 270-275.
8. Wculek, S.K., et
al., Dendritic cells in cancer immunology and immunotherapy. Nature Reviews
Immunology, 2020. 20(1): p. 7-24.
9. Del Prete, A., et al., Functional role of
dendritic cell subsets in cancer progression and clinical implications.
International journal of molecular sciences, 2020. 21(11): p. 3930.
10. Merad, M., et al., The dendritic cell
lineage: ontogeny and function of dendritic cells and their subsets in the
steady state and the inflamed setting. Annual review of immunology, 2013.
31(1): p. 563-604.
11. Plantinga, M., et al., Conventional and
monocyte-derived CD11b+ dendritic cells initiate and maintain T helper 2
cell-mediated immunity to house dust mite allergen. Immunity, 2013. 38(2): p.
322-335.
12. Kossaï, M., et al.,
Ovarian cancer: a heterogeneous disease. Pathobiology, 2018. 85(1-2): p. 41-49.
13. Reid, B.M., J.B. Permuth,
and T.A. Sellers, Epidemiology of ovarian cancer: a review. Cancer biology
& medicine, 2017. 14(1): p. 9.
14. Matulonis, U.A., et al., Ovarian cancer.
Nature reviews Disease primers, 2016. 2(1): p. 1-22.
15. McCluggage, W.G., Morphological subtypes of
ovarian carcinoma: a review with emphasis on new developments and pathogenesis.
Pathology-Journal of the RCPA, 2011. 43(5): p. 420-432.
16. Sankaranarayanan, R. and J. Ferlay, Worldwide burden of gynaecological
cancer: the size of the problem. Best practice & research Clinical
obstetrics & gynaecology, 2006. 20(2): p.
207-225.
17. Witkowski, L., et al., The influence of
clinical and genetic factors on patient outcome in small cell carcinoma of the
ovary, hypercalcemic type. Gynecologic oncology, 2016. 141(3): p. 454-460.
18. Veras, E., et al., Cystic and adenofibromatous clear cell carcinomas of the ovary:
distinctive tumors that differ in their pathogenesis and behavior: a
clinicopathologic analysis of 122 cases. The American journal of surgical
pathology, 2009. 33(6): p. 844-853.
19. Kindelberger, D.W., et al., Intraepithelial
carcinoma of the fimbria and pelvic serous carcinoma: evidence for a causal
relationship. The American journal of surgical pathology, 2007. 31(2): p.
161-169.
20. Li, J., et al., Tubal origin of ‘ovarian’low-grade serous carcinoma. Modern Pathology, 2011.
24(11): p. 1488-1499.
21. Gerhard, G.M., et al., Tumor-infiltrating
dendritic cell states are conserved across solid human cancers. Journal of
Experimental Medicine, 2021. 218(1).
22. Lynch, H.T., et al., Hereditary ovarian
carcinoma: heterogeneity, molecular genetics, pathology, and management.
Molecular oncology, 2009. 3(2): p. 97-137.
23. Ford, D., et al., Genetic heterogeneity and
penetrance analysis of the BRCA1 and BRCA2 genes in breast cancer families. The
American Journal of Human Genetics, 1998. 62(3): p. 676-689.
24. Beesley, J., et al., Association between
single-nucleotide polymorphisms in hormone metabolism and DNA repair genes and
epithelial ovarian cancer: results from two Australian studies and an
additional validation set. Cancer Epidemiology Biomarkers & Prevention,
2007. 16(12): p. 2557-2565.
25. Reid, S.D., G. Penna, and L. Adorini, The control of T cell responses by dendritic cell
subsets. Current opinion in immunology, 2000. 12(1): p. 114-121.
26. Lynch, H.T., C. Bewtra,
and J.F. Lynch, Familial peritoneal ovarian carcinomatosis: a new clinical
entity? Medical Hypotheses, 1986. 21(2): p. 171-177.
27. Zhang, X., et al., Dendritic cell vaccines in
ovarian cancer. Frontiers in Immunology, 2021. 11: p. 613773.
28. Eisenthal, A., et
al., Expression of dendritic cells in ovarian tumors correlates with clinical
outcome in patients with ovarian cancer. Human pathology, 2001. 32(8): p.
803-807.
29. Caro, A.A., et al., Dendritic cell vaccines:
A promising approach in the fight against ovarian cancer. Cancers, 2022.
14(16): p. 4037.
30. Fitzgerald, K.A. and J.C. Kagan, Toll-like
receptors and the control of immunity. Cell, 2020. 180(6): p. 1044-1066.
31. Ozkan, E. and F. Bakar-Ates, The trinity of
matrix metalloproteinases, inflammation, and cancer: A literature review of
recent updates. Anti-Inflammatory & Anti-Allergy Agents in Medicinal
Chemistry (Formerly Current Medicinal Chemistry-Anti-Inflammatory and
Anti-Allergy Agents), 2020. 19(3): p. 206-221.
32. Qu, C., et al., Role of CCR8 and other
chemokine pathways in the migration of monocyte-derived dendritic cells to
lymph nodes. The Journal of experimental medicine, 2004. 200(10): p. 1231-1241.
33. Sarivalasis, A., et
al., Cell therapies in ovarian cancer. Therapeutic Advances in Medical
Oncology, 2021. 13: p. 17588359211008399.
34. Murgaski, A., et
al., Efficacy of CD40 agonists is mediated by distinct cDC
subsets and subverted by suppressive macrophages. Cancer Research, 2022.
82(20): p. 3785-3801.
35. Guilliams, M., et al., Dendritic cells,
monocytes and macrophages: a unified nomenclature based on ontogeny. Nature
Reviews Immunology, 2014. 14(8): p. 571-578.
36. Collin, M. and V. Bigley, Human dendritic
cell subsets: an update. Immunology, 2018. 154(1): p. 3-20.
37. Maier, B., et al., A conserved dendritic-cell
regulatory program limits antitumour immunity.
Nature, 2020. 580(7802): p. 257-262.
38. Mueller, S.N., Spreading the load: Antigen
transfer between migratory and lymph node‐resident dendritic cells promotes
T‐cell priming. European Journal of Immunology, 2017. 47(10): p. 1798-1801.
39. Van Willigen, W.W., et al., Dendritic cell
cancer therapy: vaccinating the right patient at the right time. Frontiers in
Immunology, 2018. 9: p. 2265.
40. Liu, K. and M.C. Nussenzweig,
Origin and development of dendritic cells. Immunological reviews, 2010. 234(1):
p. 45-54.
41. Ma, Y., et al., Dendritic cells in the cancer
microenvironment. Journal of Cancer, 2013. 4(1): p. 36.
42. Stoitzner, P., et
al., Inefficient presentation of tumor-derived antigen by tumor-infiltrating
dendritic cells. Cancer Immunology, Immunotherapy, 2008. 57: p. 1665-1673.
43. Flies, D.B., et al., Immune checkpoint
blockade reveals the stimulatory capacity of tumor-associated CD103+ dendritic
cells in late-stage ovarian cancer. Oncoimmunology,
2016. 5(8): p. e1185583.
44. Conrad, C., et al., Plasmacytoid dendritic
cells promote immunosuppression in ovarian cancer via ICOS costimulation
of Foxp3+ T-regulatory cells. Cancer research, 2012. 72(20): p. 5240-5249.
45. Anguille, S., et
al., Clinical use of dendritic cells for cancer therapy. The lancet oncology,
2014. 15(7): p. e257-e267.
46. Huang, C.-T., et al., Insulin-like growth
factors inhibit dendritic cell-mediated anti-tumor immunity through regulating
ERK1/2 phosphorylation and p38 dephosphorylation. Cancer letters, 2015. 359(1):
p. 117-126.
47. Somri-Gannam, L.,
et al., IGF1R axis inhibition restores dendritic cell antitumor response in
ovarian cancer. Translational Oncology, 2020. 13(8): p. 100790.
48. Goyne, H.E. and M.J. Cannon, Dendritic cell
vaccination, immune regulation, and clinical outcomes in ovarian cancer.
Frontiers in immunology, 2013. 4: p. 382.
49. Tanyi, J.L., et al., Personalized cancer
vaccine effectively mobilizes antitumor T cell immunity in ovarian cancer.
Science translational medicine, 2018. 10(436): p. eaao5931.
50. Odunsi, K., et al., Vaccination with an
NY-ESO-1 peptide of HLA class I/II specificities induces integrated humoral and
T cell responses in ovarian cancer. Proceedings of the National Academy of
Sciences, 2007. 104(31): p. 12837-12842.
51. Aurisicchio, L. and
G. Ciliberto, Genetic cancer vaccines: current status and perspectives. Expert
opinion on biological therapy, 2012. 12(8): p. 1043-1058.
52. Truxova, I., et
al., Rationale for the combination of dendritic cell-based vaccination
approaches with chemotherapy agents. International Review of Cell and Molecular
Biology, 2017. 330: p. 115-156.
53. Duwa, R., J.-H. Jeong, and S. Yook,
Immunotherapeutic strategies for the treatment of ovarian cancer: current
status and future direction. Journal of Industrial and Engineering Chemistry,
2021. 94: p. 62-77.
54. An, D., S. Banerjee, and J.-M. Lee, Recent
advancements of antiangiogenic combination therapies in ovarian cancer. Cancer
treatment reviews, 2021. 98: p. 102224.
55. Deng, L., et al., STING-dependent cytosolic
DNA sensing promotes radiation-induced type I interferon-dependent antitumor
immunity in immunogenic tumors. Immunity, 2014. 41(5): p. 843-852.
56. Mastelic-Gavillet,
B., et al., Quantitative and qualitative impairments in dendritic cell subsets
of patients with ovarian or prostate cancer. European Journal of Cancer, 2020.
135: p. 173-182.
57. Guo, C., et al., Therapeutic cancer vaccines:
past, present, and future. Advances in cancer research, 2013. 119: p. 421-475.
58. Wirth, T.C., J.T. Harty, and V.P. Badovinac,
Modulating numbers and phenotype of CD8+ T cells in secondary immune responses.
European journal of immunology, 2010. 40(7): p. 1916-1926.
59. Fucikova, J., et
al., Induction of tolerance and immunity by dendritic cells: mechanisms and
clinical applications. Frontiers in immunology, 2019. 10: p. 2393.
60. Dhodapkar, M.V., et al., Induction of
antigen-specific immunity with a vaccine targeting NY-ESO-1 to the dendritic
cell receptor DEC-205. Science translational medicine, 2014. 6(232): p.
232ra51-232ra51.
61. Zhou, Y., et al., Vaccine efficacy against
primary and metastatic cancer with in vitro-generated CD103+ conventional
dendritic cells. Journal for immunotherapy of cancer, 2020. 8(1).
62. Vazquez, J., et al., Identification of unique
clusters of T, dendritic, and innate lymphoid cells in
the peritoneal fluid of ovarian cancer patients. American Journal of
Reproductive Immunology, 2020. 84(3): p. e13284.
63. Zhang, W., et al., Phase I/II clinical trial
of a Wilms’ tumor 1-targeted dendritic cell vaccination-based immunotherapy in
patients with advanced cancer. Cancer Immunology, Immunotherapy, 2019. 68: p.
121-130.
64. Mitri, Z., T.
Constantine, and R. O′ Regan, The HER2 receptor in breast cancer:
pathophysiology, clinical use, and new advances in therapy. Chemotherapy
research and practice, 2012. 2012(1): p. 743193.
65. Gulley, J.L., et al., A pilot study to
evaluate the safety and clinical outcomes of vaccination with recombinant
CEA-MUC-1-TRICOM (PANVAC) poxviral-based vaccines in patients with metastatic
carcinoma. Clinical cancer research: an official journal of the American
Association for Cancer Research, 2008. 14(10): p. 3060.
66. Fard, A.A. and A. Mahmoodzadeh,
Unraveling the Progression of Colon Cancer Pathogenesis Through Epigenetic
Alterations and Genetic Pathways. Cureus, 2024.
16(5).