Alzheimer’s disease
and glioblastoma: a comprehensive review of epidemiological and translational
medicine
Ali
Najafizadeh 1a, Elahe Bakhshalipour 1a, Maryam Gholamniya
Foumani 2, Majid Mirmazloumi 3, Mohsen Safi Samgh Abadi 4,
Zohreh Teymori 5*
1 School
of Paramedicine Sciences, Guilan University of Medical Sciences, Langarud, Iran
2 School
of Nursing and Midwifery, Guilan University of Medical Sciences, Rasht, Iran
3 Guilan
University of Medical Sciences, Rasht, Iran
4 Qazvin
University of Medical Sciences, Qazvin, Iran
5 Department
of Psychiatry, Shafa Hospital, School of Medicine, Guilan University of Medical
Sciences, Rasht, Iran
Corresponding Author: Zohreh
Teymori
* Email: Teymori.z@gmail.com
a These
authors contributed equally to this work
Abstract
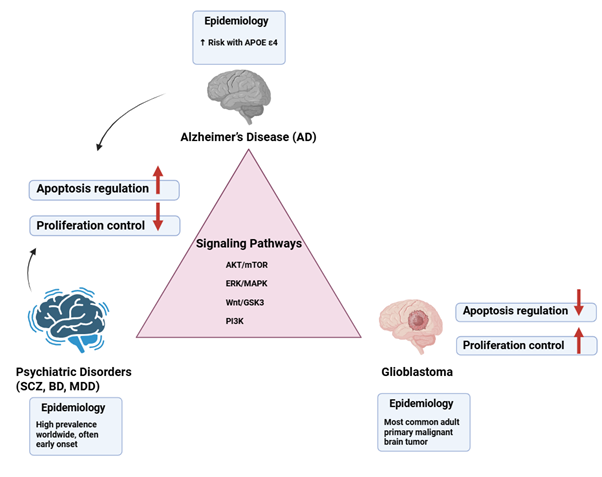
Alzheimer's disease (AD), neuropsychiatric disorders, and glioblastoma
represent distinct yet interconnected conditions characterized by overlapping
molecular, cellular, and genetic mechanisms. Epidemiological studies reveal an
inverse comorbidity between AD and glioblastoma, while psychiatric disorders
may influence glioblastoma susceptibility through genetic, cellular, and
pharmacological factors. Consequently, numerous critical signaling pathways,
such as protein kinase B/mammalian target of rapamycin (AKT/mTOR),
extracellular signal-regulated kinase / mitogen-activated protein Kinase
(ERK/MAPK), wingless-related integration Site/glycogen synthase kinase 3
(Wnt/GSK3), and phosphoinositide 3-kinase (PI3K), exhibit dysregulation across
these conditions, affecting apoptosis, proliferation, and synaptic function. In
addition, genetic risk factors, including apolipoprotein E epsilon 4 (APOE ε4)
in AD and tumor protein 53 (TP53), phosphatase and tensin homolog (PTEN), and isocitrate
dehydrogenase 1 and 2 (IDH1/2) in glioblastoma, play a role in shaping
divergent disease trajectories. Neuroinflammatory processes, epigenetic
changes, and interactions between neurons and glia further clarify
susceptibility patterns. From a therapeutic perspective, repurposing
psychiatric medications that target common molecular pathways and implementing
epigenetic or gene-based interventions offer promising avenues for integrated
treatment strategies. This review aims to synthesize the current understanding
of the epidemiological, molecular, cellular, and genetic intersections among
AD, psychiatric disorders, and glioblastoma.
Keywords: Alzheimer’s disease (AD), Glioblastoma, Neuropsychiatric disorders, Genetic
risk factors
Graphical abstract
Introduction
Alzheimer's disease (AD) and glioblastoma
constitute two of the most severe neurological conditions, exhibiting markedly
contrasting clinical and pathological features. AD is a progressive
neurodegenerative disorder marked by cognitive impairment, synaptic
dysfunction, the accumulation of amyloid-β (Aβ) plaques, tau protein
hyperphosphorylation, and extensive neuronal degeneration (1, 2). Glioblastoma, conversely, represents the most aggressive form of
primary brain tumor found in adults, characterized by unregulated cellular
growth, widespread invasion, the formation of new blood vessels, and
significant resistance to standard treatment methods (3, 4). Although both conditions impact the same organ, the human brain, their
pathophysiological mechanisms are remarkably distinct. AD is characterized by
excessive cell death, whereas glioblastoma is marked by uncontrolled cellular
survival and proliferation. The convergence of neurodegeneration and
oncogenesis has garnered increasing scientific attention over the last twenty
years. Epidemiological research has indicated an inverse comorbidity between AD
and various cancer types, including glioblastoma, suggesting that individuals
with AD may experience a lower risk of developing cancer, and conversely (5, 6). This paradox has prompted essential inquiries into the molecular and
cellular mechanisms that dictate cell fate, apoptosis, proliferation, and
immune surveillance within the central nervous system (CNS). Recent studies
have started to investigate the molecular factors contributing to this inverse
relationship. Proteins like peptidyl-prolyl cis-trans isomerase
NIMA-interacting 1 (Pin1) and p53, along with pathways such as ERK/MAPK and
PI3K/AKT, seem to be inversely regulated in AD and glioblastoma (7, 8). For instance, Pin1 is reduced in AD, which contributes to the
hyperphosphorylation of tau and subsequent neurodegeneration. In contrast, it
is frequently overexpressed in glioblastoma, facilitating the proliferation of
tumor cells (9) . In a similar vein, the activity of p53, which is usually heightened in AD
and contributes to apoptosis, is frequently rendered inactive in glioblastoma,
thereby facilitating malignant proliferation. Gaining insight into these
molecular interconnections not only illuminates the biological distinctions
between neurodegeneration and tumorigenesis but also paves the way for
potential therapeutic interventions. Furthermore, the context of cellular and
microenvironmental factors is crucial in both conditions. Microglia, the CNS's
resident immune cells, are involved in the pathology of AD through persistent
neuroinflammation and impaired clearance of Aβ (10). In glioblastoma, microglia and tumor-associated macrophages (TAMs) may
be utilized to facilitate tumor proliferation, angiogenesis, and the
suppression of immune responses (11). Exploring the interplay of neuroinflammation, oxidative stress, and
immune surveillance in the context of neurodegeneration and tumorigenesis is
crucial for comprehending disease progression. This review seeks to deliver an in-depth
examination of the relationships among AD, various neurodegenerative and
psychiatric conditions, and glioblastoma. In this manner, we emphasize
epidemiological data, molecular and cellular processes, genetic
predispositions, and therapeutic implications.
Epidemiology:
Comorbidity Patterns of AD, Neuropsychiatric Disorders, and Glioblastoma
Epidemiological studies consistently demonstrate an inverse relationship
in comorbidity between AD and glioblastoma. Extensive cohort and registry-based
research indicates that individuals with AD have a lower likelihood of
developing glioblastoma when compared to the general population (5, 12). This inverse relationship is posited to arise from intrinsic
differences in cellular destiny, like neurodegeneration, which is characterized
by apoptosis, synaptic loss, and neuronal susceptibility, whereas gliomagenesis
necessitates the avoidance of apoptosis and unchecked proliferation (8). Consequently, biological pathways that facilitate neurodegeneration in
AD may concurrently establish an unfavorable environment for the initiation of
tumors. The epidemiological context becomes increasingly intricate when
psychiatric disorders are taken into account. Numerous population-based studies
indicate that individuals diagnosed with schizophrenia, and to a lesser degree,
bipolar disorder (BD), exhibit a lower incidence of brain tumors, such as
glioblastoma (13). The protective effect has been linked to genetic variations that
enhance apoptosis, modifications in cell cycle regulation, and the prolonged
administration of psychotropic drugs that possess anti-proliferative
characteristics (14, 15). In comparison, major depressive disorder (MDD) is more reliably linked
to the risk of systemic cancer, although the evidence pertaining specifically
to glioblastoma is still scarce (16). When considered collectively, these results suggest a continuum in
which both neurodegenerative diseases, like AD, and specific neurodevelopmental
or psychiatric conditions, such as schizophrenia, might possess shared
protective molecular mechanisms against glioblastoma. In the case of AD, the
excessive activation of apoptotic and degenerative pathways diminishes neuronal
survival while simultaneously restricting neoplastic transformation. Likewise,
in schizophrenia, impaired neurodevelopment and synaptic pruning could lead to
a decrease in glial proliferative capacity. Notably, pharmacological treatments
for psychiatric conditions, including lithium, antipsychotics, and selective
serotonin reuptake inhibitors (SSRIs), further complicate this dynamic by providing
independent anti-glioma effects (15, 17). The integration of these epidemiological insights indicates that
neurodegenerative and psychiatric disorders, although clinically distinct, may
intersect in their contribution to glioblastoma risk by means of a balance
among genetic, cellular, and pharmacological factors.
Age and Gender Factors
Aging is the most significant risk factor for both AD and glioblastoma;
however, its biological effects differ markedly. In the case of AD, aging
exacerbates neuronal susceptibility due to mitochondrial dysfunction, oxidative
stress, impaired autophagy, and the progressive accumulation of Aβ and
hyperphosphorylated tau (18). Aging neurons and glial cells play a role in creating a persistent
pro-inflammatory atmosphere, referred to as inflammaging, which intensifies
neurodegeneration and cognitive deterioration (19). In contrast, in the case of glioblastoma, the aging process makes
individuals more susceptible to tumor development not by causing neuronal loss
but rather by enhancing genomic instability, disrupting DNA repair mechanisms,
and diminishing immune surveillance (20). Glioblastoma cells take advantage of age-related alterations by
obtaining mutations in the telomerase reverse transcriptase (TERT) promoter and
undergoing epigenetic reprogramming, which allows them to circumvent
replicative senescence (21). Therefore, although aging promotes apoptosis and synaptic degeneration
in AD, it concurrently contributes to cellular immortality and oncogenic
processes in glioblastoma. Additionally, gender represents a significant factor
influencing the susceptibility and progression of both AD and glioblastoma.
Notably, women are disproportionately impacted by AD, comprising nearly
two-thirds of the patient population, a discrepancy that cannot be fully
accounted for by differences in life expectancy (22). Postmenopausal estrogen deficiency is associated with increased amyloid
deposition, enhanced tau hyperphosphorylation, and diminished synaptic
resilience, underscoring the neuroprotective functions of sex hormones (23). Conversely, glioblastoma demonstrates a greater prevalence and less
favorable prognosis in males, with epidemiological research reporting
male-to-female ratios between 1.3:1 and 1.6:1 (24). Preclinical studies indicate that androgens may facilitate the
proliferation of glioma cells, whereas estrogens appear to exert protective
effects by inhibiting cellular proliferation (25). Moreover, sex-specific variations in immune responses, microglial
activation, and epigenetic regulation have been identified as contributing
factors in the pathogenesis of both AD and glioblastoma (26). Psychiatric disorders add complexity to the relationship between aging
and gender concerning the risks of AD and glioblastoma. Schizophrenia and BD
generally present during early adulthood, occurring many years before the peak
incidence of AD and glioblastoma. This indicates that neurodevelopmental
changes occurring in early life may interact with aging processes in later
stages of life (27). Interestingly, the epidemiology of mental health disorders exhibits
sex-specific trends; for example, schizophrenia tends to be more frequent and
severe among men, whereas mood disorders are more commonly observed in women (28). These variations may interact with the gender-specific risk profiles
associated with AD and glioblastoma. For instance, the neuroprotective
properties of estrogen could partly account for the reduced incidence of
glioblastoma observed in females, while also modulating susceptibility to
affective disorders and AD following menopause. Conversely, males diagnosed
with schizophrenia might benefit from protective effects against glioblastoma
attributable to genetic variants that promote apoptosis, despite their
inherently higher baseline risk of glioblastoma relative to females, which is
influenced by biological sex differences (14). Table 1 represents the comorbidity patterns of AD, neuropsychiatric
disorders, and glioblastoma.
Table 1. Age, sex, and genetic factors influence
the risk of AD, psychiatric disorders, and glioblastoma. Additionally,
psychiatric conditions and treatments such as lithium or antipsychotics may
reduce glioblastoma risk by promoting apoptosis.
|
Factor
|
AD
|
Glioblastoma
|
Psychiatric
Disorders
|
Connection
/ Mechanism
|
|
Age
|
Increases neuronal vulnerability, apoptosis, oxidative stress,
amyloid & tau accumulation
|
Increases genomic instability, TERT mutations, and immune evasion
|
Early-life onset (schizophrenia/bipolar) may intersect with aging
pathways later
|
Aging drives degeneration in AD but tumorigenesis in glioblastoma
|
|
Gender
|
Women > Men estrogen protective
|
Men > Women; androgens promote proliferation
|
Schizophrenia: men > women
mood disorders: women > men
|
Sex hormones modulate neurodegeneration, tumor proliferation, and
psychiatric risk
|
|
AD vs Glioblastoma
|
Neurodegeneration
|
Oncogenesis
|
–
|
Inverse comorbidity: apoptotic pathways protect against glioblastoma
|
|
Schizophrenia / Bipolar
|
–
|
Reduced glioblastoma risk
|
Neurodevelopmental alterations, apoptosis-promoting genetic
variants
|
Protective effect against glioblastoma despite baseline gender
risk
|
|
Major Depression
|
–
|
Data limited
|
Higher systemic cancer risk
|
glioblastoma-specific link unclear
|
|
Pharmacology
|
–
|
Anti-glioma effects
|
Lithium, antipsychotics, SSRIs
|
Drugs modify glioblastoma risk independently of disease
|
Environmental
and Lifestyle Factors
Environmental factors significantly influence the risk of developing
glioblastoma. Among these, ionizing radiation is the sole environmental risk
factor that has been conclusively validated through clinical and
epidemiological research (29). Individuals who have undergone cranial
irradiation, particularly during childhood, exhibit a significantly heightened
lifetime risk of developing glioma. Additionally, occupational exposure to
petrochemicals, pesticides, and heavy metals has been linked to an increased
incidence of glioblastoma; however, the supporting evidence for these
associations is variable and not consistently conclusive (30). Significantly, urban air pollution and
fine particulate matter (PM2.5) are increasingly acknowledged as factors that
contribute to oxidative DNA damage and neuroinflammation, processes that are
pertinent to both neurodegeneration in Alzheimer's disease and tumorigenesis in
glioblastoma (31). Lifestyle factors exert a significant
impact on the risk and progression of AD. Specifically, tobacco use and
excessive alcohol intake contribute to increased oxidative stress, vascular
injury, and amyloid plaque accumulation, thereby hastening cognitive
deterioration (32). In contrast, adherence to Mediterranean
or DASH dietary patterns, which are abundant in antioxidants, omega-3 fatty
acids, and polyphenols, has been associated with a decreased risk of AD and
enhanced cognitive resilience (33). Engagement in physical activity
facilitates neurogenesis, mitigates neuroinflammatory processes, and improves
cerebral perfusion, thereby collectively contributing to the postponement of
disease onset (34). These lifestyle modifications highlight
the potential of non-pharmacological interventions to mitigate the risk of AD
and to influence common biological pathways associated with glioblastoma.
Individuals diagnosed with psychiatric disorders are disproportionately
subjected to detrimental lifestyle and environmental risk factors, such as
tobacco use, unhealthy dietary habits, physical inactivity, and elevated
prevalence of substance abuse (35). These behaviors not only elevate the
overall risk of cancer but may also contribute to gliomagenesis through
mechanisms such as the induction of DNA damage, the promotion of chronic
inflammation, and the disruption of immune surveillance (36). Nonetheless, individuals within
psychiatric populations often undergo prolonged treatment with psychotropic
medications, several of which (such as lithium, antipsychotics, and SSRIs) have
demonstrated anti-glioma effects in preclinical studies (37). This interaction indicates that
although lifestyle factors may elevate the risk of cancer, pharmacological
interventions could mitigate these dangers, leading to the seemingly
contradictory observation of a lower incidence of glioblastoma among psychiatric
populations (Figure 1).
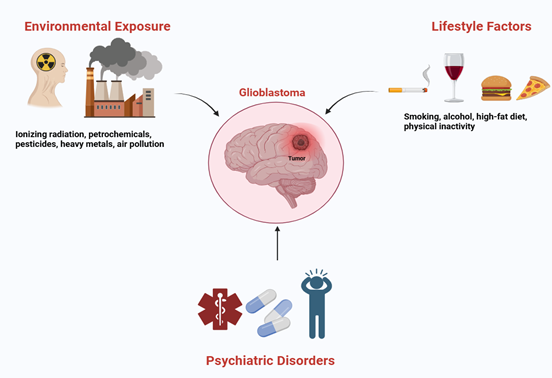
Figure 1. Environmental and lifestyle factors,
including radiation, pollution, diet, and physical activity, influence
Alzheimer’s, psychiatric disorders, and glioblastoma, with some medications
offering protective effects against tumor development.
Molecular
Mechanisms: Shared and Inverse Pathways Between AD, Psychiatric Disorders, and Glioblastoma
At the molecular level, AD, psychiatric disorders, and glioblastoma
demonstrate intricate interactions that could elucidate the observed patterns
of comorbidity. While
AD is marked by neuronal degeneration and disrupted synaptic signaling,
glioblastoma is characterized by unchecked cellular growth and invasion. Psychiatric
disorders, such as schizophrenia, BD, and MDD, frequently exhibit changes in
neurotransmitter systems, inflammation, and regulation of the cell cycle, which
may influence vulnerability to tumor development. The key molecular pathways
relevant to these subjects are outlined, including Pin1 and protein folding,
the p53 pathway, ERK/MAPK, PI3K/AKT, Wnt/GSK3 signaling, as well as
neuroinflammation and cytokine networks, as presented in (Table 2). Pin1
is a distinctive enzyme that facilitates the isomerization of phosphorylated
serine/threonine-proline motifs, thereby affecting protein structure and
functionality. Pin1
plays a crucial role in regulating protein conformation and signaling that is
dependent on phosphorylation (38). In AD, the expression of Pin1 is
reduced, resulting in the hyperphosphorylation and aggregation of tau protein.
In contrast, glioblastoma often exhibits elevated levels of Pin1, which
facilitates cell cycle advancement, cellular proliferation, and resistance to
programmed cell death. This opposing regulation of Pin1 indicates a potential
molecular mechanism that may explain the lower incidence of glioblastoma seen
in patients with AD (39, 40). Furthermore, the tumor suppressor
protein p53 serves as a vital regulator of the cell cycle, DNA repair
mechanisms, and apoptosis, commonly known as the guardian of the genome. In AD,
p53 is often upregulated in response to oxidative stress and DNA damage, which
promotes neuronal apoptosis and plays a role in neurodegeneration. Increased
levels of p53 can worsen synaptic dysfunction, resulting in cognitive decline.
Additionally, the interaction between p53 and Aβ, as well as tau proteins,
amplifies cellular stress responses, activating apoptotic pathways and causing
mitochondrial dysfunction in neurons, which are fundamental to the pathology of
AD (41). In glioblastoma, p53 frequently
undergoes mutation or functional inactivation, enabling cells to evade
apoptosis and proliferate without restraint. The loss of p53 function in this
malignancy is correlated with genomic instability, increased tumor aggressiveness,
and therapeutic resistance. This differential regulation of p53 in AD versus
glioblastoma may partly account for the observed inverse comorbidity: neurons
in AD are susceptible to p53-mediated apoptosis, whereas glial tumor cells in
glioblastoma circumvent this pathway. From a therapeutic perspective,
modulation of p53 signaling is under investigation both to mitigate excessive
neuronal loss in AD and to reinstate tumor suppressive mechanisms in
glioblastoma, underscoring the protein’s dual role in neurodegeneration and
oncogenesis (42). In addition, the ERK/MAPK and PI3K/AKT
pathways are central regulators of cell survival, proliferation,
differentiation, and synaptic plasticity. In AD, these pathways are often
disrupted: ERK/MAPK signaling shows impaired activation, which compromises
synaptic function and long-term potentiation, leading to memory deficits.
Similarly, PI3K/AKT activity is frequently reduced in AD neurons, favoring
apoptosis through activation of pro-apoptotic proteins like Bax and inhibition
of survival signals, contributing to progressive neurodegeneration.
Dysregulation of these pathways also affects tau phosphorylation, enhancing the
formation of neurofibrillary tangles (43). In glioblastoma, these signaling
pathways are markedly hyperactivated, leading to uncontrolled tumor
proliferation, increased invasiveness, and resistance to therapeutic
interventions. Dysregulated ERK/MAPK and PI3K/AKT signaling pathways facilitate
cell cycle progression, angiogenesis, and the avoidance of apoptosis, thereby
constituting critical targets for therapeutic strategies in glioblastoma
management. Similarly, psychiatric disorders exhibit differential modulation of
these pathways; for instance, schizophrenia and BD frequently demonstrate
altered AKT activity, which may impact neuronal survival and, notably, could
influence susceptibility to tumor development. Consequently, ERK/MAPK and
PI3K/AKT signaling pathways represent a molecular nexus that links neurodegenerative
processes, oncogenesis, and psychiatric conditions (44). Furthermore, the tumor suppressor
protein p53 serves as a vital regulator of the cell cycle, DNA repair
mechanisms, and apoptosis, commonly known as the guardian of the genome. In AD,
p53 is often upregulated in response to oxidative stress and DNA damage, which
promotes neuronal apoptosis and plays a role in neurodegeneration. Increased
levels of p53 can worsen synaptic dysfunction, resulting in cognitive decline.
Additionally, the interaction between p53 and Aβ, as well as tau proteins,
amplifies cellular stress responses, activating apoptotic pathways and causing
mitochondrial dysfunction in neurons, which are fundamental to the pathology of
AD. (45). In glioblastoma, the Wnt signaling
pathway is often excessively activated, thereby facilitating the maintenance of
stem cell-like properties, enhancing invasiveness, and contributing to
chemoresistance within tumor cells. Dysregulated Wnt/β-catenin signaling
drives cellular proliferation and migration, positioning it as a critical
factor in the malignancy of glioblastoma. Notably, inhibition of GSK3 in
glioblastoma has been shown to suppress tumor growth, whereas in AD,
hyperactivation of GSK3 is implicated in disease pathogenesis, demonstrating an
inverse molecular relationship between these conditions. Furthermore,
modulation of Wnt/GSK3 signaling pathways in psychiatric disorders may affect
neurodevelopmental mechanisms and cellular resilience, underscoring the role of
these pathways as shared regulators in neurodegeneration, oncogenesis, and
mental health disorders (46). Neuroinflammation constitutes a
fundamental aspect of AD as well as numerous psychiatric disorders,
predominantly mediated by microglial activation, astrocyte dysfunction, and
aberrant cytokine signaling. In the context of AD, persistent microglial activation
triggered by amyloid-beta plaques and tau protein aggregates results in the
prolonged secretion of pro-inflammatory cytokines, including interleukin-1 beta
(IL-1β), interleukin-6 (IL-6), and tumor necrosis factor-alpha
(TNF-α). This sustained inflammatory response exacerbates neuronal damage
and impairs synaptic function. Concurrently, astrocytes, which typically
support neuronal homeostasis, undergo a phenotypic transformation to a reactive
state under inflammatory conditions, thereby intensifying neurodegenerative
processes. Analogously, psychiatric conditions such as MDD and schizophrenia
are frequently characterized by low-grade systemic and CNS inflammation, with
dysregulated cytokine expression contributing to disturbances in mood
regulation, cognitive impairments, and potentially altered cellular
proliferation mechanisms. In glioblastoma, the tumor microenvironment
manipulates the immune system to facilitate tumor progression. Glioblastoma
cells promote immunosuppressive phenotypes in microglia and infiltrating
macrophages, concurrently secreting cytokines such as IL-6 and transforming
growth factor-beta (TGF-β), which contribute to tumor proliferation,
angiogenesis, and invasion. Notably, cytokines implicated in neuronal death in
AD may paradoxically enhance tumor survival in glioblastoma, underscoring an
inverse relationship between neurodegeneration and cancer. This dual
functionality of inflammatory signaling pathways indicates that precise
modulation of cytokine networks holds therapeutic promise: attenuating chronic
neuroinflammation in AD and psychiatric disorders may confer neuroprotection,
whereas targeting tumor-promoting inflammation in glioblastoma could inhibit
tumor growth. A comprehensive understanding of the context-dependent roles of cytokines
is therefore essential for the development of effective interventions
addressing both neurodegenerative and oncological conditions (2).
Table 2. Key signaling pathways, including Pin1, p53, ERK/AKT, Wnt/GSK3, and
neuroinflammation, show both shared and opposing dysregulation across these
conditions. In AD, downregulation often drives neurodegeneration, while in
glioblastoma, hyperactivation promotes proliferation and therapy resistance.
Psychiatric disorders show subtler, modulatory changes affecting synaptic
function and resilience. These patterns highlight how the same molecular
mechanisms can lead to divergent disease outcomes depending on context.
|
Molecular
Pathway
|
AD
|
Glioblastoma
|
Psychiatric
Disorders
|
|
Pin1 / Protein Folding
|
Downregulated → tau hyperphosphorylation and aggregation
|
Overexpressed → proliferation, resistance to apoptosis
|
Limited data; possible indirect effects on synaptic regulation
|
|
p53 Pathway
|
Upregulated → oxidative stress response, neuronal
apoptosis, cognitive decline
|
Mutated/inactivated → evasion of apoptosis, genomic
instability, tumor aggressiveness
|
Altered regulation in some disorders may influence stress
response and apoptosis
|
|
ERK/MAPK and PI3K/AKT
|
Reduced activity → impaired LTP, memory loss, increased
apoptosis
|
Hyperactivated → tumor growth, angiogenesis, therapy
resistance
|
Altered AKT signaling in schizophrenia/BD → impacts
neuronal survival, possible tumor susceptibility
|
|
Wnt/β-catenin and GSK3
|
Downregulated Wnt, hyperactive GSK3 → tau pathology,
neurodegeneration
|
Hyperactive Wnt/β-catenin → stemness, invasion,
chemoresistance
|
Dysregulated Wnt/GSK3 → neurodevelopmental abnormalities,
synaptic dysfunction
|
|
Neuroinflammation and Cytokines
|
Chronic microglial activation → IL-1β, IL-6,
TNF-α → synaptic loss, neuronal death
|
Glioblastoma cells co-opt microglia/macrophages → IL-6,
TGF-β → proliferation, immunosuppression
|
Low-grade inflammation (IL-6, TNF-α) → mood/cognitive
dysfunction, altered resilience
|
Cellular Mechanisms: Microglia,
Neuronal-Glial Interactions, and Tumorigenesis
The cellular microenvironment is critically influential in shaping the
progression of neurodegenerative diseases and tumorigenic processes. In
conditions such as Alzheimer's disease, psychiatric disorders, and
glioblastoma, the interplay among neurons, glial cells, and immune cells
constitutes a fundamental mechanism underlying disease development and may
account for the epidemiologically observed inverse comorbidity patterns. Microglia,
the intrinsic immune cells of the CNS, exhibit a dualistic function in the
contexts of neurodegeneration and tumorigenesis. AD, persistent microglial
activation contributes to chronic neuroinflammation, impaired clearance of
Aβ plaques, and synaptic dysfunction. Activated microglia secrete
pro-inflammatory cytokines, including IL-1β, IL-6, and TNF-α, as well
as reactive oxygen species (ROS) and nitric oxide, which collectively
facilitate neuronal apoptosis and exacerbate cognitive decline. This sustained
inflammatory milieu disrupts neurotrophic support and modifies neuron-glia
interactions, thereby driving progressive neurodegeneration. Conversely, in
glioblastoma, microglia and TAMs are co-opted into a tumor-supportive phenotype
characterized by the secretion of factors that enhance cellular proliferation,
angiogenesis, and immunosuppression. This tumor-promoting microglial phenotype
stands in contrast to their pro-apoptotic role observed in AD. By modulating
the local cytokine environment, glioblastoma cells exploit microglial
plasticity to promote tumor invasion and resistance to therapy. This functional
duality of microglia provides a mechanistic basis for the inverse
epidemiological correlation observed between neurodegenerative disorders and
gliomagenesis (47). Equally significant, astrocytes play a
crucial role in maintaining synaptic homeostasis, offering metabolic support to
neurons, and regulating inflammation within the CNS. In the context of AD,
astrocytes exhibit a reactive state, characterized by hypertrophy and the
release of inflammatory mediators that worsen neuronal damage. This reactive
gliosis is implicated in the formation of plaques, loss of synapses, and disruption
of neurovascular coupling. Furthermore, dysfunctional astrocytes in various
psychiatric disorders, such as schizophrenia and BD, also disrupt
neurotransmitter homeostasis and inflammatory responses, potentially
influencing the risk of neurodegeneration or tumor development. In
glioblastoma, astrocytes engage with tumor cells through gap junctions,
cytokine signaling, and the remodeling of the extracellular matrix, thereby
promoting tumor invasion and resistance to therapeutic interventions (48). Tumor-associated astrocytes facilitate
the preservation of glioma stem cells and promote angiogenesis by releasing
trophic factors and influencing immune responses. The differing functions of
astrocytes, acting as pro-degenerative agents in AD compared to their
pro-tumorigenic role in glioblastoma, underscore the capacity of glial
plasticity to result in varied cellular consequences within the CNS (49). Besides, interactions between neurons
and glia are crucial for maintaining homeostasis in the CNS, facilitating
synaptic plasticity, and enabling repair mechanisms. In the context of AD, the
deterioration of synaptic integrity, the reduction of neurotrophic support, and
the disruption of signaling pathways involving glutamate and other
neurotransmitters compromise neuron-glia communication, which in turn
contributes to neurodegenerative processes. The disruption of these
interactions has a detrimental impact on astrocytes, microglia, and
oligodendrocytes, thereby exacerbating cognitive deficits and neuronal loss (50). Glioblastoma cells manipulate the
communication between neurons and glia to facilitate tumor growth. For
instance, glioma cells react to neuronal activity via neuroligin-3 signaling
and utilize synaptic-like structures to boost their proliferation. This appropriation
of neuronal mechanisms stands in stark contrast to the degenerative signaling
seen in AD and specific psychiatric conditions, underscoring how analogous
cellular signals can result in either cell death or unchecked proliferation,
contingent upon the context. More importantly, Oligodendrocytes play a crucial
role in providing myelin and metabolic support to axons, which is essential for
the proper functioning of neurons. In AD, the dysfunction of oligodendrocytes
and the resulting demyelination led to impaired axonal conduction and cognitive
decline. Likewise, psychiatric conditions such as schizophrenia are linked to
changes in oligodendrocyte density, abnormalities in myelin, and compromised
integrity of white matter, all of which may affect neuronal vulnerability and
cellular resilience. In the case of glioblastoma, tumor cells engage with
oligodendrocyte progenitor cells (OPCs) to alter the extracellular matrix and
establish a microenvironment that promotes invasion. This interaction
underscores a context-dependent shift in which oligodendrocytes can either
enhance CNS function or be repurposed to aid in tumor growth (49). Cellular senescence represents a
defining characteristic of aging and plays a significant role in both
neurodegeneration and the biology of cancer (51). In AD, neurons display signs of DNA
damage-induced senescence and apoptotic signaling, which contribute to cell
loss and cognitive deterioration. Factors associated with the
senescence-associated secretory phenotype (SASP), such as pro-inflammatory
cytokines, exacerbate neuroinflammation (52). In glioblastoma, tumor cells circumvent
senescence by activating telomerase, inactivating p53, and increasing the
levels of anti-apoptotic proteins, which allows for unrestrained proliferation. Psychiatric
disorders might influence apoptotic thresholds through oxidative stress and
modified signaling pathways, thereby indirectly impacting tumor vulnerability. This contrast
between cell death in neurodegeneration and the evasion of senescence in cancer
elucidates the inverse epidemiological patterns observed (53).
Genetics and
Risk Loci: Shared and Divergent Genetic Mechanisms in AD, Psychiatric
Disorders, and Glioblastoma
Genetic elements play a crucial role in determining the vulnerability,
advancement, and clinical results of AD and glioblastoma (54). Both common and distinct genetic
mechanisms could elucidate epidemiological findings, including the inverse
comorbidity observed between neurodegenerative diseases and specific cancers,
while also offering potential molecular targets for therapeutic intervention (55). The following provides an elucidation
of the risk-associated genes implicated in these disorders:
AD Risk Genes
Genetic predisposition is crucial in AD, with risk genes impacting not
only neurodegeneration but also the brain's microenvironment, which may in turn
influence tumor biology, including glioblastoma (56). One of the primary genetic risk factors
is APOE ε4; the ε4 allele of apolipoprotein E represents the most
critical risk element for late-onset AD, as it enhances Aβ aggregation,
hinders its clearance, and facilitates tau hyperphosphorylation (57). In addition to amyloidopathy, APOE
ε4 influences lipid metabolism, neuroinflammation, and synaptic
plasticity. Microglia from individuals carrying the APOE ε4 allele display
a pro-inflammatory phenotype, secreting cytokines like IL-1β and TNF-α, which promote
neuronal apoptosis (58). Although this environment promotes
neurodegeneration, the identical pro-apoptotic signaling may render glial
progenitors less conducive to tumor initiation, which partially elucidates the
noted inverse comorbidity between AD and glioblastoma. Furthermore, mutations
in APP and presenilins 1 and 2 modify γ-secretase activity, leading to an
increased production of the neurotoxic Aβ42 isoform (59). This disparity encourages synaptic
degradation, oxidative stress, and dysfunction of mitochondria. Notably,
persistent oxidative stress can initiate DNA damage responses that might
inhibit unchecked proliferation in glial cells, thereby possibly restricting
tumor development. Additionally, Microglial and Immune-Related Genes, such as triggering
receptor expressed on myeloid cells 2 (TREM2), Clusterin (CLU), and cluster of differentiation
33 (CD33), play crucial roles in regulating microglial phagocytosis, lipid
metabolism, and inflammatory signaling (60, 61). Loss-of-function mutations in TREM2
diminish the ability of microglia to clear Aβ, thereby worsening the
pathology; however, they may concurrently improve glial monitoring against
malignant transformation. CLU plays a role in regulating apoptosis and
interactions with the extracellular matrix, which affects neuronal survival and
the microenvironment conducive to glioblastoma growth. Additionally, epigenetic
modulators, including genes that affect DNA methylation and histone
modifications (for instance, DNA (Cytosine-5)-Methyltransferase 1 (DNMT1) and histone
deacetylases (HDACs), also play a role in the risk of AD (62). Epigenetic dysregulation has the
potential to modify neuronal gene expression, diminish proliferation, and
sustain a cellular condition that is resistant to malignant transformation.
Glioblastoma Risk
Genes
Glioblastoma represents a highly aggressive primary brain tumor
influenced by a complex interaction of genetic, epigenetic, and
microenvironmental elements. Gaining insight into susceptibility genes aids in
clarifying the reasons certain pathways overlap with neurodegenerative and
psychiatric conditions. For instance, TP53 serves as a vital tumor suppressor
that governs DNA repair, apoptosis, and the regulation of cell cycle checkpoints
(63). Loss-of-function mutations in TP53
frequently occur in glioblastoma, especially in secondary glioblastoma that
develops from lower-grade gliomas. The presence of mutant p53 enables tumor
cells to escape apoptosis even in the presence of DNA damage, which facilitates unchecked
proliferation. Notably, the hyperactivation of p53 in neurons is associated
with apoptosis in AD, underscoring the context-dependent duality of p53
signaling that serves a protective role against malignancy in glial cells while
being pro-apoptotic in neurons (64, 65). Also, PTEN negatively regulates the
PI3K/AKT pathway, a central driver of cell survival and proliferation (66). The impairment of PTEN function in
glioblastoma results in the hyperactivation of AKT, which facilitates metabolic
reprogramming, angiogenesis, and therapeutic resistance. In relation to
neurodegeneration, the modulation of PTEN influences neuronal survival and the
guidance of axons. Consequently, the dysfunction of PTEN may play a role in
tumorigenesis while also intersecting with pathways associated with
neuropsychiatric disorders (67). Furthermore, the amplification of epidermal
growth factor receptor (EGFR) and mutations such as EGFRvIII contribute to
oncogenic signaling via the MAPK, PI3K/AKT, and signal transducer and activator
of transcription 3 (STAT3) pathways (68). This facilitates cellular
proliferation, migration, and resistance to programmed cell death. Notably,
dysregulated EGFR signaling has been associated with psychiatric conditions
such as schizophrenia and BD, potentially through its impact on
neurodevelopmental mechanisms and synaptic plasticity, indicating the existence
of a common molecular framework (69). An additional important aspect to
highlight is that neurofibromin 1 (NF1) functions as a negative regulator of rat
sarcoma (RAS) signaling. Loss-of-function mutations in the NF1 gene result in
increased activity of the RAS/MAPK pathway, thereby facilitating glial cell
proliferation and tumor development (70). Dysfunction of NF1 also impacts
neuronal differentiation and synaptic plasticity, thereby connecting
neurodevelopmental processes with an increased susceptibility to tumor
formation.
Shared
Molecular Pathways Between AD and Glioblastoma
AD and glioblastoma exhibit distinct pathological outcomes; they share
several molecular pathways, underscoring the complexity of context-dependent
effects. For example, the Pin1 pathway functions differently in these
conditions: in AD, Pin1 facilitates the correction of tau protein misfolding
and inhibits the formation of neurofibrillary tangles, whereas in glioblastoma,
Pin1 contributes to the stabilization of oncogenic proteins such as cyclin D1
and Akt, thereby promoting cell cycle progression and cellular proliferation (71). The dual function demonstrates that
pathway activity may serve a protective role in neurons while being oncogenic
in glial cells. Furthermore, p53 upholds genomic integrity by triggering cell
cycle arrest or apoptosis when faced with DNA damage. In the context of
neurons, the activation of p53 plays a role in apoptosis and neurodegeneration
associated with AD. Conversely, in glioblastoma, mutations in TP53 compromise
this checkpoint, enabling cells to escape apoptosis. The shared regulation of
p53 underscores a pivotal point where identical molecular mechanisms yield
contrasting outcomes in various cell types (42, 72). Furthermore, the PI3K/AKT/mTOR pathway
plays a crucial role. This pathway regulates cellular growth, survival, and
metabolic processes. In AD, the excessive activation of mTOR leads to tau
hyperphosphorylation and hinders autophagy (44). In glioblastoma, the hyperactivity of
the PI3K/AKT/mTOR pathway leads to accelerated cell proliferation, metabolic
alterations, and increased resistance to programmed cell death. Consequently,
pharmacological agents that inhibit mTOR may possess a dual therapeutic effect:
they could mitigate AD pathology while concurrently inhibiting tumor growth in
glioblastoma. Additionally, the ERK/MAPK signaling pathway plays a crucial role
in regulating cell proliferation, differentiation, and responses to stress. In
the context of AD, its dysregulation contributes to tau phosphorylation,
oxidative stress, and neuronal cell death (73). In glioblastoma, the identical pathway
promotes both proliferation and invasion. Variations that depend on context,
such as the availability of different cofactors and the signaling from upstream
receptors, dictate whether ERK/MAPK activity results in degeneration or
proliferation. Additionally, regarding oxidative stress and responses to DNA
damage, both AD and glioblastoma are associated with the generation of ROS, yet
the outcomes are distinct. In AD, oxidative stress triggers neuronal apoptosis
and mitochondrial dysfunction. Conversely, in glioblastoma, tumor cells
frequently utilize ROS signaling to facilitate DNA repair, angiogenesis, and
survival in hypoxic environments (74). Furthermore, autophagy is compromised
in AD, resulting in the buildup of Aβ and tau aggregates. In contrast, in
glioblastoma, autophagy plays a crucial role in promoting the survival of tumor
cells during metabolic stress (75). The therapeutic modulation of autophagy
can thus be customized to either avert neurodegeneration or suppress tumor
growth.
Neuropsychiatric
Disorders and Glioblastoma: Molecular and Cellular Intersections
Neuropsychiatric disorders, such as schizophrenia, BD, and MDD, exhibit
intricate associations with glioblastoma across molecular, cellular, and
epidemiological dimensions. While population-based research indicates a
typically reduced occurrence of glioblastoma among individuals with severe
psychiatric conditions, emerging mechanistic understanding is being derived
from genetic, signaling, and cellular investigations (76, 77) (Figure 2). Here are some crucial
examples of it:
1.
Schizophrenia and Glioblastoma
Schizophrenia is associated with disturbances in dopaminergic,
glutamatergic, and GABAergic signaling, along with atypical neurodevelopment
and synaptic plasticity. Candidate genes like disrupted in schizophrenia 1 (DISC1),
neuregulin 1 (NRG1), and AKT Serine/Threonine Kinase 1 (AKT1) are involved in
the regulation of neuronal proliferation, migration, and apoptosis. The dysregulation
of AKT/mTOR and ERK/MAPK pathways may diminish the proliferation of glial
precursors, which could restrict the reservoir of cells that are susceptible to
malignant transformation (78). Furthermore, variations in genes that
control oxidative stress and DNA repair processes may provide neuroprotection
while concurrently decreasing the likelihood of tumor development. In the context of
schizophrenia, there are noticeable changes in neuron-glia interactions, which
encompass a decrease in oligodendrocyte density, compromised myelination, and
disturbances in synaptic pruning (79). These structural deficiencies may
undermine the specific environment necessary for glioblastoma cell
proliferation, thereby diminishing tumor initiation. Prolonged exposure to
antipsychotic medications additionally influences glial function, encompassing
microglial apoptosis and the proliferation of astrocytes, which contributes an
extra dimension of protection (63). Antipsychotic medications, especially
those that act as dopamine receptor antagonists, exhibit anti-proliferative
properties in glioma cell lines, which include the suppression of cell cycle
progression and the promotion of apoptosis. Consequently, prolonged treatment
may provide further defense against the onset of glioblastoma. Subsequent research could aim
to explore the potential of these drugs as supplementary anti-glioma treatments
(37).
2. BD and Glioblastoma
BD is linked to disrupted intracellular signaling, oxidative stress,
mitochondrial impairment, and neuroinflammation. Significant molecular
pathways, such as glycogen synthase kinase 3 beta (GSK3β), Wnt/β-catenin,
and PI3K/AKT, frequently exhibit dysregulation in both BD and glioblastoma.
Lithium, a widely used mood stabilizer, acts by inhibiting GSK3β and
modulating Wnt signaling, which are pathways that are excessively active in
glioblastoma, consequently restricting glial proliferation and invasiveness (80). Patients with BD often display modified
neuron-glia ratios, reduced oligodendrocyte density, and compromised
myelination. Such cellular irregularities may restrict the assistance for tumor
initiation and growth (81). Furthermore, persistent inflammation
and oxidative stress can trigger DNA damage responses that encourage the
apoptosis of cells that may initiate tumors. The inhibition of GSK3β by lithium also
enhances neurotrophic signaling through brain-derived neurotrophic factor (BDNF),
potentially safeguarding neurons while restricting the survival of glioma cells
(82). Other mood stabilizers, such as
valproate, modify epigenetic landscapes through histone deacetylase inhibition,
which may further reduce glioblastoma susceptibility. Collectively, genetic
predisposition, cellular microenvironment, and pharmacological interventions
converge to reduce glioblastoma risk in BD patients (83).
3. MDD and Glioblastoma
MDD is defined by persistent systemic inflammation, dysregulation of the
HPA axis, and oxidative stress. Increased levels of pro-inflammatory cytokines
such as IL-6, TNF-α, and IL-1β influence microglial activation and
apoptosis, which may restrict glial proliferation within the CNS (84). Genetic variants associated with
apoptosis, DNA repair mechanisms, and mitochondrial function may play a role in
the pathophysiology of MDD as well as in decreased susceptibility to
glioblastoma. Persistent neuroinflammation observed in MDD facilitates glial
cell apoptosis and disrupts astrocytic support for neuronal function (85). The altered microenvironment may
exhibit decreased permissiveness for glioblastoma progression, thereby limiting
the available niche for tumor initiation. Additionally, stress-induced
increases in glucocorticoid levels further regulate the functions of microglia
and astrocytes, impacting the proliferation and apoptosis of pre-neoplastic
glial cells (86). Antidepressant agents, such as SSRIs
and SNRIs, have exhibited anti-glioma properties in vitro by promoting
apoptosis, autophagy, and oxidative stress within glioma cells. The interplay
between neuroinflammatory alterations associated with disease pathology and the
pharmacodynamic actions of these medications may act synergistically to
mitigate the risk of glioblastoma (87).
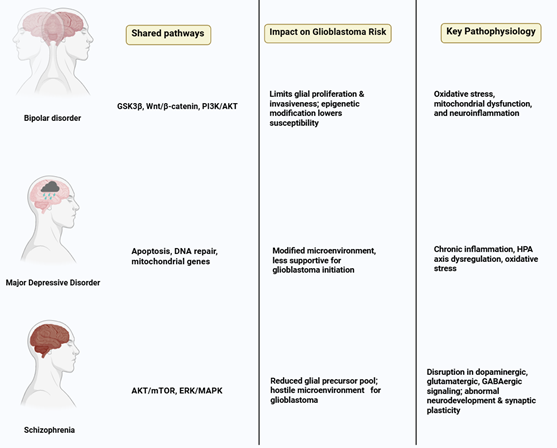
Figure 2. Shared molecular pathways, including
AKT/mTOR, ERK/MAPK, GSK3β/Wnt, and PI3K/AKT, alongside alterations in
oxidative stress, mitochondrial function, and neuroinflammatory signaling,
collectively contribute to a neural microenvironment less permissive to
glioblastoma initiation and progression. These intersecting mechanisms
highlight how psychiatric pathophysiology and pharmacological modulation may
influence glial proliferation and tumor susceptibility.
Implications
for Therapy and Prevention
The convergence of neuropsychiatric conditions and glioblastoma opens
novel avenues for developing innovative treatments and preventive strategies.
These approaches seek not only to address distinct disease states but also to
exploit inverse comorbidity trends to advance personalized medicine. Critical
molecular pathways, including PI3K/AKT/mTOR and Wnt/β-catenin signaling,
play pivotal roles in regulating cell growth, viability, and invasiveness in
glioblastoma. Therapies targeting these pathways have demonstrated potential to
slow tumor growth and enhance patient outcomes. For instance, lithium-mediated
suppression of GSK3β has shown antitumor effects in preclinical
glioblastoma studies. Repurposing established psychiatric medications offers a
compelling strategy for glioblastoma management. Antipsychotic drugs, such as
clozapine and aripiprazole, have been observed to curb glioma cell growth and
trigger apoptosis. Likewise, SSRIs, such as sertraline, exhibit anti-glioma
properties by modulating pathways associated with cell proliferation and tumor
invasion. These findings suggest that prolonged use of psychiatric medications
may impact glioblastoma risk and provide new therapeutic possibilities.
Conclusion
AD, psychiatric conditions, and glioblastoma are distinct disorders
with unique symptoms, yet they share underlying molecular, cellular, and
genetic connections. Population-based studies suggest an intriguing negative
correlation between AD and GBM, while psychiatric disorders may alter GBM risk
through genetic, cellular, and drug-related mechanisms. At the molecular level,
disrupted signaling networks, such as those involving PI3K/AKT/mTOR, ERK/MAPK,
and Wnt/GSK3β, drive neurodegeneration or tumor development in contrasting
ways. Proteins like Pin1 and p53, along with neuroinflammatory and synaptic
processes, reveal shared regulatory frameworks that lead to either neuronal
loss or excessive glial growth. Cellular interactions, including microglial
responses, astrocyte functions, neuron-glia dynamics, and synaptic adaptations,
help explain these differing disease outcomes. Genetic studies point to the
role of risk variants, epigenetic alterations, and overlapping signaling genes
in shaping disease predisposition. In AD, genes that heighten neuronal
susceptibility may create a brain environment less favorable to GBM. Similarly,
genes linked to psychiatric conditions influence tumor formation by modulating
cell survival, apoptosis, and oxidative stress. Drugs used for psychiatric and
neurodegenerative disorders, such as antipsychotics, mood stabilizers, and
SSRIs, may lower GBM risk by affecting glial cell behavior. From a treatment
perspective, these insights pave the way for tailored therapeutic strategies.
By targeting shared signaling pathways, managing neuroinflammation, or using
epigenetic and genetic approaches, new treatments could emerge. Repurposing
existing medications or designing therapies that address multiple targets may
benefit both brain disorders and GBM management. In conclusion, the complex
interplay of neurodegenerative, psychiatric, and tumor-related processes calls
for an integrated approach combining molecular science, genetics, neuroscience,
and pharmacology. Future research leveraging multi-omics, long-term population
studies, and experimental models will be vital to uncover the basis of these
disease connections and develop precise interventions that bridge these
seemingly distinct conditions.
Acknowledgments
We extend our
sincere gratitude to all individuals who contributed to the development of this
narrative review article.
Author
contribution
AN and EB authored the primary manuscript and developed the
accompanying figures and tables. ZT revised and finalized the
manuscript. MGF and MM contributed to the authorship of certain
sections of the manuscript. All authors have reviewed and approved the
final revised version of the manuscript.
Funding
There is no funding.
Conflicts of interest
There are no conflicts of interest.
References
1. Bloom GS.
Amyloid-β and tau: the trigger and bullet in Alzheimer disease
pathogenesis. JAMA neurology. 2014;71(4):505-8.
2. Cai Y, Liu J, Wang B,
Sun M, Yang H. Microglia in the Neuroinflammatory Pathogenesis of Alzheimer's
Disease and Related Therapeutic Targets. Front Immunol. 2022;13:856376.
3. Stupp R, Mason WP, Van
Den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus
concomitant and adjuvant temozolomide for glioblastoma. New England journal of
medicine. 2005;352(10):987-96.
4. Tan AC, Ashley DM,
López GY, Malinzak M, Friedman HS, Khasraw M. Management of glioblastoma: State
of the art and future directions. CA: a cancer journal for clinicians.
2020;70(4):299-312.
5. Driver JA, Beiser A, Au
R, Kreger BE, Splansky GL, Kurth T, et al. Inverse association between cancer
and Alzheimer’s disease: results from the Framingham Heart Study. Bmj.
2012;344.
6. Zhang Q, Guo S, Zhang
X, Tang S, Shao W, Han X, et al. Inverse relationship between cancer and
Alzheimer’s disease: a systemic review meta-analysis. Neurological Sciences.
2015;36(11):1987-94.
7. Liu T, Ren D, Zhu X,
Yin Z, Jin G, Zhao Z, et al. Transcriptional signaling pathways inversely
regulated in Alzheimer's disease and glioblastoma multiform. Scientific
reports. 2013;3(1):3467.
8. Driver JA, Zhou XZ, Lu
KP. Pin1 dysregulation helps to explain the inverse association between cancer
and Alzheimer's disease. Biochimica et Biophysica Acta (BBA)-General Subjects.
2015;1850(10):2069-76.
9. Seo J, Park M.
Molecular crosstalk between cancer and neurodegenerative diseases. Cell Mol
Life Sci. 2020;77(14):2659-80.
10. Abate G, Frisoni GB,
Bourdon J-C, Piccirella S, Memo M, Uberti D. The pleiotropic role of p53 in
functional/dysfunctional neurons: focus on pathogenesis and diagnosis of
Alzheimer’s disease. Alzheimer's Research & Therapy. 2020;12(1):160.
11. Andersen RS, Anand A,
Harwood DSL, Kristensen BW. Tumor-Associated Microglia and Macrophages in the
Glioblastoma Microenvironment and Their Implications for Therapy. Cancers
(Basel). 2021;13(17).
12. Zhang D-D, Ou Y-N, Fu Y,
Wang Z-B, Huang L-Y, Tan L, Yu J-T. Risk of dementia in cancer survivors: a
meta-analysis of population-based cohort studies. Journal of Alzheimer’s
Disease. 2022;89(1):367-80.
13. Catts V, Catts S,
O’toole B, Frost A. Cancer incidence in patients with schizophrenia and their
first‐degree relatives–a meta‐analysis. Acta Psychiatrica
Scandinavica. 2008;117(5):323-36.
14. Tabarés-Seisdedos R,
Rubenstein JL. Inverse cancer comorbidity: a serendipitous opportunity to gain
insight into CNS disorders. Nature Reviews Neuroscience. 2013;14(4):293-304.
15. Jia N, Li Z, Li X, Jin
M, Liu Y, Cui X, et al. Long-term effects of antipsychotics on mortality in
patients with schizophrenia: a systematic review and meta-analysis. Brazilian
Journal of Psychiatry. 2022;44:664-73.
16. Oerlemans ME, van den
Akker M, Schuurman AG, Kellen E, Buntinx F. A meta-analysis on depression and
subsequent cancer risk. Clinical Practice and Epidemiology in Mental Health.
2007;3(1):29.
17. Morselli E, Galluzzi L,
Kepp O, Vicencio J-M, Criollo A, Maiuri MC, Kroemer G. Anti-and pro-tumor
functions of autophagy. Biochimica et Biophysica Acta (BBA)-Molecular Cell
Research. 2009;1793(9):1524-32.
18. Mao P, Reddy PH. Aging
and amyloid beta-induced oxidative DNA damage and mitochondrial dysfunction in
Alzheimer's disease: implications for early intervention and therapeutics.
Biochim Biophys Acta. 2011;1812(11):1359-70.
19. Franceschi C, Campisi J.
Chronic inflammation (inflammaging) and its potential contribution to
age-associated diseases. Journals of Gerontology Series A: Biomedical Sciences
and Medical Sciences. 2014;69(Suppl_1):S4-S9.
20. Ohgaki H, Kleihues P.
The definition of primary and secondary glioblastoma. Clinical cancer research.
2013;19(4):764-72.
21. Vinagre J, Almeida A,
Pópulo H, Batista R, Lyra J, Pinto V, et al. Frequency of TERT promoter
mutations in human cancers. Nature communications. 2013;4(1):2185.
22. Beam CR, Kaneshiro C,
Jang JY, Reynolds CA, Pedersen NL, Gatz M. Differences between women and men in
incidence rates of dementia and Alzheimer’s disease. Journal of Alzheimer’s
disease. 2018;64(4):1077-83.
23. Price BR, Walker KA,
Eissman JM, Suryadevara V, Sime LN, Hohman TJ, Gordon MN. Sex differences and
the role of estrogens in the immunological underpinnings of Alzheimer's
disease. Alzheimers Dement (N Y). 2025;11(3):e70139.
24. Ostrom QT, Gittleman H,
Liao P, Rouse C, Chen Y, Dowling J, et al. CBTRUS statistical report: primary
brain and central nervous system tumors diagnosed in the United States in
2007–2011. Neuro-oncology. 2014;16(suppl_4):iv1-iv63.
25. Yang W, Warrington NM,
Taylor SJ, Whitmire P, Carrasco E, Singleton KW, et al. Sex differences in GBM
revealed by analysis of patient imaging, transcriptome, and survival data.
Science translational medicine. 2019;11(473):eaao5253.
26. Villa A, Gelosa P,
Castiglioni L, Cimino M, Rizzi N, Pepe G, et al. Sex-specific features of
microglia from adult mice. Cell reports. 2018;23(12):3501-11.
27. Insel TR. Rethinking
schizophrenia. Nature. 2010;468(7321):187-93.
28. Kessler RC, Bromet EJ.
The epidemiology of depression across cultures. Annual review of public health.
2013;34(1):119-38.
29. Fardone E. A new
application of microbeam radiation therapy (MRT) on the treatment of epilepsy
and brain disorders: Université de Grenoble; 2013.
30. Wrensch M, Minn Y, Chew
T, Bondy M, Berger MS. Epidemiology of primary brain tumors: current concepts
and review of the literature. Neuro-oncology. 2002;4(4):278-99.
31. Machens A, Dralle H. Age
disparities in referrals to specialist surgical care for papillary thyroid
cancer. Eur J Surg Oncol. 2009;35(12):1312-7.
32. Durazzo TC, Mattsson N,
Weiner MW. Smoking and increased Alzheimer's disease risk: a review of
potential mechanisms. Alzheimers Dement. 2014;10(3 Suppl):S122-45.
33. Holmes P, Collins D,
Calmels C. Electroencephalographic functional equivalence during observation of
action. J Sports Sci. 2006;24(6):605-16.
34. Erickson KI, Voss MW,
Prakash RS, Basak C, Szabo A, Chaddock L, et al. Exercise training increases
size of hippocampus and improves memory. Proc Natl Acad Sci U S A.
2011;108(7):3017-22.
35. Schotland SV. The Fight
Over the Juvenile Giant: Contesting Growth in the 1930s. Pediatrics.
2019;144(2).
36. Stutzmann F, Cauchi S,
Durand E, Calvacanti-Proença C, Pigeyre M, Hartikainen AL, et al. Common
genetic variation near MC4R is associated with eating behaviour patterns in
European populations. Int J Obes (Lond). 2009;33(3):373-8.
37. Kamarudin MNA, Parhar I.
Emerging therapeutic potential of anti-psychotic drugs in the management of
human glioma: A comprehensive review. Oncotarget. 2019;10(39):3952-77.
38. Kimura T, Tsutsumi K,
Taoka M, Saito T, Masuda-Suzukake M, Ishiguro K, et al. Isomerase Pin1
stimulates dephosphorylation of tau protein at cyclin-dependent kinase
(Cdk5)-dependent Alzheimer phosphorylation sites. J Biol Chem.
2013;288(11):7968-77.
39. Butterfield DA, Abdul
HM, Opii W, Newman SF, Joshi G, Ansari MA, Sultana R. Pin1 in Alzheimer's
disease. J Neurochem. 2006;98(6):1697-706.
40. Bulbarelli A, Lonati E,
Cazzaniga E, Gregori M, Masserini M. Pin1 affects Tau phosphorylation in
response to Abeta oligomers. Mol Cell Neurosci. 2009;42(1):75-80.
41. Wolfrum P, Fietz A,
Schnichels S, Hurst J. The function of p53 and its role in Alzheimer's and
Parkinson's disease compared to age-related macular degeneration. Front
Neurosci. 2022;16:1029473.
42. Zhang Y, Dube C, Gibert
M, Jr., Cruickshanks N, Wang B, Coughlan M, et al. The p53 Pathway in
Glioblastoma. Cancers (Basel). 2018;10(9).
43. Chen MJ, Ramesha S,
Weinstock LD, Gao T, Ping L, Xiao H, et al. Extracellular signal-regulated
kinase regulates microglial immune responses in Alzheimer's disease. J Neurosci
Res. 2021;99(6):1704-21.
44. Razani E,
Pourbagheri-Sigaroodi A, Safaroghli-Azar A, Zoghi A, Shanaki-Bavarsad M,
Bashash D. The PI3K/Akt signaling axis in Alzheimer's disease: a valuable
target to stimulate or suppress? Cell Stress Chaperones. 2021;26(6):871-87.
45. Sai Varshini M,
Aishwarya Reddy R, Thaggikuppe Krishnamurthy P. Unlocking hope: GSK-3
inhibitors and Wnt pathway activation in Alzheimer's therapy. J Drug Target.
2024;32(8):909-17.
46. Robertson FL, O'Duibhir
E, Gangoso E, Bressan RB, Bulstrode H, Marqués-Torrejón M, et al. Elevated
FOXG1 in glioblastoma stem cells cooperates with Wnt/β-catenin to induce
exit from quiescence. Cell Rep. 2023;42(6):112561.
47. Di Nunno V, Aprile M,
Gatto L, Tosoni A, Ranieri L, Bartolini S, Franceschi E. Tumor Microenvironment
in Gliomas: A Treatment Hurdle or an Opportunity to Grab? Cancers (Basel).
2023;15(4).
48. Gómez-Gonzalo M,
Martin-Fernandez M, Martínez-Murillo R, Mederos S, Hernández-Vivanco A, Jamison
S, et al. Neuron-astrocyte signaling is preserved in the aging brain. Glia.
2017;65(4):569-80.
49. Buruiană A, Florian
Ș I, Florian AI, Timiș TL, Mihu CM, Miclăuș M, et al. The
Roles of miRNA in Glioblastoma Tumor Cell Communication: Diplomatic and
Aggressive Negotiations. Int J Mol Sci. 2020;21(6).
50. Smaili SS, Ureshino RP,
Rodrigues L, Rocha KK, Carvalho JT, Oseki KT, et al. The role of mitochondrial
function in glutamate-dependent metabolism in neuronal cells. Curr Pharm Des.
2011;17(35):3865-77.
51. Alshaebi F, Sciortino A,
Kayed R. The Role of Glial Cell Senescence in Alzheimer's Disease. J Neurochem.
2025;169(3):e70051.
52. Li R, Li Y, Zuo H, Pei
G, Huang S, Hou Y. Alzheimer's Amyloid-β Accelerates Cell Senescence and
Suppresses SIRT1 in Human Neural Stem Cells. Biomolecules. 2024;14(2).
53. Mandelblatt JS, Hurria
A, McDonald BC, Saykin AJ, Stern RA, VanMeter JW, et al. Cognitive effects of
cancer and its treatments at the intersection of aging: what do we know; what
do we need to know? Semin Oncol. 2013;40(6):709-25.
54. Papoff G, Presutti D,
Lalli C, Bolasco G, Santini S, Manelfi C, et al. CASP4 gene silencing in
epithelial cancer cells leads to impairment of cell migration, cell-matrix
adhesion and tissue invasion. Sci Rep. 2018;8(1):17705.
55. Hamzavi-Zarghani Z,
Yahaghi A, Matekovits L, Farmani A. Tunable mantle cloaking utilizing graphene
metasurface for terahertz sensing applications. Opt Express.
2019;27(24):34824-37.
56. Singh S, Dey D, Barik D,
Mohapatra I, Kim S, Sharma M, et al. Glioblastoma at the crossroads: current
understanding and future therapeutic horizons. Signal Transduct Target Ther.
2025;10(1):213.
57. Liu CC, Liu CC, Kanekiyo
T, Xu H, Bu G. Apolipoprotein E and Alzheimer disease: risk, mechanisms and
therapy. Nat Rev Neurol. 2013;9(2):106-18.
58. Dias D, Portugal CC,
Relvas J, Socodato R. From Genetics to Neuroinflammation: The Impact of ApoE4
on Microglial Function in Alzheimer's Disease. Cells. 2025;14(4).
59. Valdés-Rives SA,
Casique-Aguirre D, Germán-Castelán L, Velasco-Velázquez MA, González-Arenas A.
Apoptotic Signaling Pathways in Glioblastoma and Therapeutic Implications.
Biomed Res Int. 2017;2017:7403747.
60. Selvaraj NR, Nandan D,
Nair BG, Nair VA, Venugopal P, Aradhya R. Oxidative Stress and Redox Imbalance:
Common Mechanisms in Cancer Stem Cells and Neurodegenerative Diseases. Cells.
2025;14(7).
61. Li RY, Qin Q, Yang HC,
Wang YY, Mi YX, Yin YS, et al. TREM2 in the pathogenesis of AD: a lipid
metabolism regulator and potential metabolic therapeutic target. Mol
Neurodegener. 2022;17(1):40.
62. Schoch KM, Ezerskiy LA,
Morhaus MM, Bannon RN, Sauerbeck AD, Shabsovich M, et al. Acute Trem2 reduction
triggers increased microglial phagocytosis, slowing amyloid deposition in mice.
Proc Natl Acad Sci U S A. 2021;118(27).
63. Pouyan A, Ghorbanlo M,
Eslami M, Jahanshahi M, Ziaei E, Salami A, et al. Glioblastoma multiforme:
insights into pathogenesis, key signaling pathways, and therapeutic strategies.
Mol Cancer. 2025;24(1):58.
64. Noor H, Briggs NE,
McDonald KL, Holst J, Vittorio O. TP53 Mutation Is a Prognostic Factor in Lower
Grade Glioma and May Influence Chemotherapy Efficacy. Cancers (Basel).
2021;13(21).
65. Jebelli JD, Hooper C,
Garden GA, Pocock JM. Emerging roles of p53 in glial cell function in health
and disease. Glia. 2012;60(4):515-25.
66. Rascio F, Spadaccino F,
Rocchetti MT, Castellano G, Stallone G, Netti GS, Ranieri E. The Pathogenic
Role of PI3K/AKT Pathway in Cancer Onset and Drug Resistance: An Updated
Review. Cancers (Basel). 2021;13(16).
67. Bao L, Li X, Lin Z. PTEN
overexpression promotes glioblastoma death through triggering mitochondrial
division and inactivating the Akt pathway. J Recept Signal Transduct Res.
2019;39(3):215-25.
68. Uribe ML, Marrocco I,
Yarden Y. EGFR in Cancer: Signaling Mechanisms, Drugs, and Acquired Resistance.
Cancers (Basel). 2021;13(11).
69. Keshri N, Nandeesha H.
Dysregulation of Synaptic Plasticity Markers in Schizophrenia. Indian J Clin
Biochem. 2023;38(1):4-12.
70. Anastasaki C, Orozco P,
Gutmann DH. RAS and beyond: the many faces of the neurofibromatosis type 1
protein. Dis Model Mech. 2022;15(2).
71. Proctor CJ, Gray DA.
GSK3 and p53 - is there a link in Alzheimer's disease? Mol Neurodegener.
2010;5:7.
72. Farmer KM, Ghag G,
Puangmalai N, Montalbano M, Bhatt N, Kayed R. P53 aggregation, interactions
with tau, and impaired DNA damage response in Alzheimer's disease. Acta
Neuropathol Commun. 2020;8(1):132.
73. Zamanian MY, Khachatryan
LG, Heidari M, Darabi R, Golmohammadi M, Al-Aouadi RFA, Akkol EK. The
Therapeutic Potential of Flavonols in Alzheimer's Disease: Inhibiting
Amyloid-β, Oxidative Stress, and Neuroinflammation. Biofactors.
2025;51(5):e70047.
74. Kamat PK, Kalani A, Rai
S, Swarnkar S, Tota S, Nath C, Tyagi N. Mechanism of Oxidative Stress and
Synapse Dysfunction in the Pathogenesis of Alzheimer's Disease: Understanding
the Therapeutics Strategies. Mol Neurobiol. 2016;53(1):648-61.
75. Barmaki H, Nourazarian
A, Khaki-Khatibi F. Proteostasis and neurodegeneration: a closer look at
autophagy in Alzheimer's disease. Front Aging Neurosci. 2023;15:1281338.
76. Gao X, Mi Y, Guo N, Xu
H, Jiang P, Zhang R, et al. Glioma in Schizophrenia: Is the Risk Higher or
Lower? Front Cell Neurosci. 2018;12:289.
77. Yang W, Han Y, He C,
Zhong S, Ren F, Chen Z, et al. Association between psychiatric disorders and
glioma risk: evidence from Mendelian randomization analysis. BMC Cancer.
2024;24(1):118.
78. Buonanno A. The
neuregulin signaling pathway and schizophrenia: from genes to synapses and
neural circuits. Brain Res Bull. 2010;83(3-4):122-31.
79. Ermakov EA, Dmitrieva
EM, Parshukova DA, Kazantseva DV, Vasilieva AR, Smirnova LP. Oxidative
Stress-Related Mechanisms in Schizophrenia Pathogenesis and New Treatment
Perspectives. Oxid Med Cell Longev. 2021;2021:8881770.
80. Muneer A. Wnt and GSK3
Signaling Pathways in Bipolar Disorder: Clinical and Therapeutic Implications.
Clin Psychopharmacol Neurosci. 2017;15(2):100-14.
81. Gibson EM, Geraghty AC,
Monje M. Bad wrap: Myelin and myelin plasticity in health and disease. Dev
Neurobiol. 2018;78(2):123-35.
82. Kandezi N, Mohammadi M,
Ghaffari M, Gholami M, Motaghinejad M, Safari S. Novel Insight to
Neuroprotective Potential of Curcumin: A Mechanistic Review of Possible
Involvement of Mitochondrial Biogenesis and PI3/Akt/ GSK3 or PI3/Akt/CREB/BDNF
Signaling Pathways. Int J Mol Cell Med. 2020;9(1):1-32.
83. Berendsen S, Frijlink E,
Kroonen J, Spliet WGM, van Hecke W, Seute T, et al. Effects of valproic acid on
histone deacetylase inhibition in vitro and in glioblastoma patient samples.
Neurooncol Adv. 2019;1(1):vdz025.
84. Hassamal S. Chronic
stress, neuroinflammation, and depression: an overview of pathophysiological
mechanisms and emerging anti-inflammatories. Front Psychiatry. 2023;14:1130989.
85. Huang J, Pham VT, Fu S,
Huang G, Liu YG, Zheng L. Mitophagy's impacts on cancer and neurodegenerative
diseases: implications for future therapies. J Hematol Oncol. 2025;18(1):78.
86. Lin H, Liu C, Hu A,
Zhang D, Yang H, Mao Y. Understanding the immunosuppressive microenvironment of
glioma: mechanistic insights and clinical perspectives. J Hematol Oncol.
2024;17(1):31.
87. Ge Y, Cao Y, Wang Q,
Wang Y, Ma W. Impact of antidepressant use on survival outcomes in glioma
patients: A systematic review and meta-analysis. Neurooncol Adv.
2024;6(1):vdae181.