Figure 1. Phosphine functions by interfering
with the mitochondrial respiratory chain at the level of the mitochondria.
Induced cytotoxicity
caused the mitochondrial damages and oxidative stress by Aluminum phosphide; an
overview of the mechanism to the clinic
Morteza Rahbar Taramsari 1*, Hamid Mohammadi Kojidi
1*
1 Razi Clinical Research Development Unit, Razi Hospital, Guilan
University of Medical Sciences, Rasht, Iran
Corresponding Authors: Morteza Rahbar
Taramsari * Email: rahbar_m46@yahoo.com
Hamid Mohammadi Kojidi * Email: h_mohammadi8778@yahoo.com
Abstract
Aluminum phosphide (AlP) is a significant fumigant and a notable,
highly effective pesticide for both indoor and outdoor use. Analytical tests
like the gas chromatographic method in post-mortem specimens and survivors have
been developed to assess the quantity of phosphine and to differentiate between
ZnP and AlP poisoning, even if clinical history can usually aid in making the
final diagnosis. In this way, it is demonstrated that mitochondrial failure
caused ALP to create reactive oxygen species (ROS). As a result of red blood
cell hemolysis, decreased ATP synthesis, and the activation of apoptosis in
cardiomyocytes brought on by ROS generation, different problems eventually
develop. Since cardiomyocytes are the cells that are most significantly
affected by ALP, using the right therapeutic methods to get the cells working
again will prolong patient survival. Correspondingly, Phosphine's ability to
inhibit cytochrome c oxidase has been demonstrated in vitro. It seems
improbable that this interaction is the main driver of its toxicity, though.
ALP poisoning may cause the most damage to the mitochondria, which might lead
to poor ATP synthesis, metabolic shutdown, and multiorgan dysfunction (MOD).
Additionally, due to an impairment in electron flow, there may be free radical
formation and damage, which could also result in MOD. Rats and insects have
shown signs of ALP-induced toxicity brought on by reactive oxygen species. A
similar mechanism might potentially be present in people and help fill in the
gap in the pathophysiology of ALP poisoning. Cellular poisoning, oxidative
stress, cholinesterase inhibition, circulatory failure, cardiotoxicity,
gastrointestinal and pulmonary toxicity, hepatic damage, neurological toxicity,
electrolyte imbalance, and general metabolic disturbances are just a few of the
many effects caused by metal phosphides.
In this review article, we discuss the association of cytotoxicity,
mitochondrial damage, and oxidative stress by Aluminum phosphide.
Keywords: Aluminum phosphide, Cytotoxicity, Mitochondrial damage, Oxidative
stress
Introduction
Aluminum
phosphide (AlP) is an essential fumigant, a commendable and very superb outside
and indoor insecticide and rodenticide, extensively bought and used because the
1940s. AlP is effortlessly reachable as pellets or a pill formulated and
offered in porous baggage in a stable form, underneath change names such as
Phostoxin, Quickphos Phosphume Phostek, Bhostoxin, Quickphos, Alphos, and
Celphos (can launch 1 g PH3). It is utilized in growing international locations
in suicide tries. AlP is handy in pesticide demands as an affordable grain
rodenticide (1).
High
viable houses are the purpose for the significance of its availability. The
residences are close to perfect toxicity species, no longer concerning the
viability of the seeds, leaving little remains on meal grains, being
lower-priced and notably formulation. Moisture in the air mixed with aluminum
phosphide, makes phosphine gas, the direct lively poison.
AlP
is a frequent substance used in some Asian and European locations as a frequent
approach to suicide. AlP, domestically known as the "rice pill", is
extensively used to defend rice. In factories, most publicity entails
swallowing suicide or unintended exposure, especially thru meals by using
farmers and pores and skin exposure, which hardly ever reasons extreme systemic
toxicity (2).
The
Chemistry of AlP
AlP
is normally accessible in stable structure positioned in blister packs,
commonly synthesized as darkish brown/gray or yellow crystals that include 44%
aluminum carbonate and 56% AlP. Absorption of phosphine gas, with odorless and
colorless properties, is speedy thru mucosal and pores and skin contact,
inhalation and ingestion due to the formation of diphosphines. After ingestion,
which is the most frequent way of exposure, a small volume of zinc phosphide
attains to the kidneys and liver and is hydrolyzed stilly in the tissues. Zinc
phosphide is synthesized by way of a mixture of phosphorus and zinc.
Due
to the production of diphosphines, the odorless and colorless phosphine gas is
quickly absorbed by mucosal and skin contact, breathing, and ingestion (3, 4). A little
amount of zinc phosphide enters the liver and kidneys after ingestion, which is
the most common exposure method and is hydrolyzed slowly in the tissues. Zinc
and phosphorus are combined to create zinc phosphide (5).
Practical
action
It
has been established that the fatal dose of AlP is approximately 0.5 g. The
simplest method of absorbing is through oral consumption. ALP gas is released
when various phosphide salts, particularly hydrochloric acid, react with
stomach contents. The cytochrome C oxidase enzyme and mitochondrial electron
transport chain are stopped after tissue absorption (6).
Phosphine
mostly binds to cytochrome oxidase, changing the hemoglobin's valences and
eventually causing protein aggregation, organ-specific cell membrane damage,
and lipid peroxidation. According to some research, a significant phosphine
level is associated with a decrease in serum cholinesterase (7). Ionic barrier disruption, and
protein degradation, induce apoptosis, nucleic acid disruption, and ultimately
cell death take place. AlP furthermore significantly lowers glutathione, a
powerful antioxidant defense molecule. Malondialdehyde (MDA), superoxide
dismutase (SOD), and catalase levels have been linked to AlP mortality.
Phosphine has a significant part in the conformational changes of oxyhemoglobin,
which can cause oxidative injury to cellular life, notably in the brain, lung,
and liver, correspondent to research on both humans and animals (8).
Clinical
indicators and diagnosis
Phosphine
vaporizes immediately after ingesting a very small quantity of an AlP tablet
due to air contact, and it disrupts several organs. The heart, digestive tract,
respiratory system, and kidneys are the primary organs impacted by the initial
exposure. Other symptoms include pulmonary edema, nausea, cyanosis, epigastric
discomfort abdominal pain, and palpitations. Other symptoms include cardiac
arrhythmias, shock, and metabolic acidosis, which are connected to myocardium
injury that has been mentioned in some cases. Preliminary signs of AlP
poisoning, such as nausea, agitation, epigastric discomfort and vomiting, and
leucopenia, are important indicators (9).
Clinical
symptoms and lab evaluation guide the use of clinical diagnostics. According to
the findings, hepatocytes were damaged and severe AlP toxicity was indicated by
increasing levels of serum glutamic pyruvic transaminase (SGPT) and glutamic
oxaloacetic transaminase (SGOT) produced metabolic acidosis (10) (Table 1).
Table 1. Clinical symptoms of ALP poisoning.
|
System poisoning |
Clinical characteristics |
|
Respiratory |
Obstructive pulmonary disease, pleural effusion, pulmonary edema,
adult respiratory distress syndrome, lung inflammation |
|
Neurological |
Headache,
acute dysfunction of the brain, weakness, ataxia, neuropathy tremor,
paraesthesias |
|
Hematological |
Disseminated intravascular coagulation, intravascular hemolysis,
methemoglobinemia |
|
Gastrointestinal |
Esophagitis,
tracheoesophageal fistula, ascites, hepatic disorders, esophageal strictures |
|
Cardiovascular |
Dysrhythmias, pericardial effusion, low blood pressure,
progressive cardiac conduction defect, ventricular dysfunction, pericarditis,
shock, myocarditis |
|
Metabolic |
Low
potassium, metabolic acidosis, low blood sugar level, hypermagnesemia, low
level of serum magnesium |
|
Renal |
Regeneration of tubular epithelium, acute renal disorder,
congestion within glomerules |
Common
symptoms include tachypnea, dyspnea, crepitations, and rhonchi. Although
pulmonary edema is frequently present, its cause may be either cardiogenic or
noncardiogenic. PH3 appears to interact with moisture in the lungs after
inhalation to create phosphoric acid, which in turn damages the alveolar
membrane. Adult respiratory distress syndrome (ARDS) cases associated with ALP
poisoning have been documented, corroborating this assertion. Those who survive
PH3 exposure also appear to experience long-term side effects such as
obstructive airway disease (11).
According
to a current, contentious study, chronic liver destruction brought on by AlP
poisoning can result in hepatotoxicity. The primary results in this regard
included centrilobular necrosis, hepatocyte nuclei being destroyed, fatty liver
alterations, and central venous congestion. Hepatocellular toxicity and acute
fulminant hepatic failure, which have been observed in some acute intoxication
cases in various studies, have also been identified to be potential causes of
death. Numerous investigations have also notified common ECG changes and
cardiovascular complications like PR and QRS interval prolongation, ST-segment
elevation that causes severe hypotension by lowering systemic venous pressure,
complete heart block due to ectopic pacemaking, and irreversible myocardial
injury, and atrial fibrillation. There are ST-T alterations and sinus
tachycardia in the first 3 to 6 hours following poisoning, which are followed
by conduction abnormalities and persistent arrhythmias in the following 6 to 12
hours (12).
The
exact cause of why liver disease frequently manifests in less severe ways is
unknown. After consuming metal phosphide, transient increases of serum alanine
aminotransferase and aspartate aminotransferase have been observed, however,
liver damage-related jaundice is far less frequent. The most common autopsy
findings include portal edema, congestion of the portal tract and central
veins, and vacuolization of hepatocytes (13).
Different
examples of myocardial damage have been documented that involve anterior wall
ischemia with RBBB (right bundle branch block), wave flattening suggesting
myocardial ischemia, and total RBBB (14).
Hematemesis,
vomiting, fistula, esophageal strictures, and epigastric discomfort are the
most prominent gastrointestinal symptoms of AlP consumption and result in upper
gastrointestinal bleeding. Various endoscopic reports have described slugged
mucosa and the destruction of the stomach and esophageal tissues (15).
Previous
research has shown that dysphagia may develop later on as a result of the
mucosa's increased slugging during ALP poisoning. There are some signs of a
necrotic and thinner stomach wall, as well as mucosa, in the stomach's fundus
wall. During oral AlP poisoning, there have been cases of spontaneous
inflammation and stomach wall burns (16). Water and electrolyte imbalances
can produce hypokalemia either as a causative factor of vomiting or as a
subsequent effect. Acute renal failure, metabolic acidosis, respiratory
alkalosis, and significant variations in calcium, phosphate, magnesium,
cortisol, and citrate levels have all been noted. Various variations in blood
glucose levels are also observed (17).
To
investigate rice tablet poisoning, the silver nitrate test is utilized since it
is a significant, straightforward, and sensitive spot examination. To detect
inhaled PH3 gas, use fresh silver nitrate solution paper. The sample color
turns black with this technique. Some sophisticated biochemical assays employ
blood or gastric aspiration samples to find phosphine. the detection of
phosphine gas in samples is the most reliable method for ALP poisoning
diagnosis. The phosphine in the bio-samples is characterized using ion
chromatographic techniques. Analytical assays were utilized to measure the
quantity of phosphine and to differentiate between ZnP and AlP poisoning in
post-mortem specimens and survivors (18).
Tissue
morphology after exposure to AlP
Numerous
studies have looked into how tissues alter morphologically after being exposed
to or poisoned with AlP. Target organs for AlP poisoning include the liver,
brain, kidneys, heart, and lungs. Additionally, microscopic examination reveals
various degrees of edema, inflammation, and congestion in bodily organs.
Analysis identified congestion, interstitial edema, hemorrhage, varying degrees
of alveolar collapse, alveolar thickening, and emphysema as the primary
histological abnormalities in the lung tissue (19).
ALP
poisoning caused significant necrosis in the liver due to the morphological
examination of the liver sample revealing vacuolar degeneration in hepatocytes
cells, central venous congestion, mononuclear infiltration, sinusoidal
dilatation, and centrilobular hemorrhagic necrosis. Portal edema, centrilobular
necrosis, nuclear fragmentation, clusters of polymorph nuclear leukocytes in
sinusoids, subcapsular hemorrhage, and macrovesicular steatosis are nonetheless
prevalent histopathologic diagnoses. In the aforementioned investigation,
sinusoidal clusters of polymorph nuclear leukocytes, nuclear hepatocytes, and
sinusoidal congestion were reported. In severe AlP poisoning, the plasma level
of renin increases after liver injury while cortisol levels fall at the upper
level. When the kidneys were examined under a microscope, abnormalities
included swelling of the epithelial cells of the proximal convoluted tubules,
glomeruli and intraparenchymal congestion, and the renal medulla (20).
Control
of poisoning
Control
of AlP poisoning should begin right away. Initially, a thorough history must be
obtained, followed as quickly as feasible by a clinical assessment. The
majority of intoxication management involves supportive measures such as
mechanical breathing, inotropic support, and fluid resuscitation. The majority
of therapy attempts, nevertheless, have not been wholly effective and
appropriate, and no definitive therapeutic has yet been presented; various
therapeutic approaches are included below (21).
Digestive
system decontamination
After
consuming AlP, various gastrointestinal symptoms including diarrhea, vomiting,
abdominal soreness, and eventually epigastric and abdominal pain, were described.
Vomiting
is a common complaint among patients, however gastric lavage with a 1/5000
potassium permanganate solution removes and/or oxidizes unabsorbed toxins.
However, during gastric lavage, caution must be exercised to avoid aspiration.
A 2 percent solution of bicarbonate can also be used to neutralize hydrochloric
acid and then stop the release of phosphine. When the bicarbonate level is
below 15 mEq/L, sodium bicarbonate must be administered intravenously at
minimum doses of 50–100 mEq every 8 hours until the bicarbonate level reaches
18–20 mEq/L (21, 22).
Administration
of sorbitol solution as a cathartic and liquid paraffin and vegetable oils as
blockers of phosphine produced from the AlP are two more therapies that have
been suggested. According to a study conducted on ALP poisoning, coconut oil
can help treat acute phosphine poisoning in people up to six hours after
exposure. It is uncertain how coconut oil and other lipids work in the
digestive system. Through the formation of a protective barrier surrounding the
stomach mucosa, dilution of stomach HCl, and decreased phosphide breakdown,
coconut oil reduces the absorption of PH3 gas. Rats poisoned with AlP had a
substantial reduction in mortality after gastric lavage with sweet almond oil,
which also decreased plasma cholinesterase levels. It has been advised to do a
wide stomach lavage while also mixing coconut oil and sodium bicarbonate
solution (23, 24).
Due
to ALP's corrosive nature and the fact that oral ingestion is the most typical
method of poisoning, GI tract symptoms are frequently the first and most
prevalent. Retrosternal burning, epigastric discomfort, and vomiting are the
early signs and symptoms following consumption. Excessive thirst, stomach pain,
and tenderness in the epigastric region are gastrointestinal symptoms that
appear with moderate to severe poisoning. Hematemesis, perhaps even large
hematemesis, is one way that ALP's esophageal corrosive action might appear.
Dysphagia in survivors may become apparent as early as 3 or 4 days after
ingesting aluminum phosphide. Esophageal strictures have afterward developed in
several cases. Trachea-esophageal fistulae have been documented in a few cases (25).
Treatment
of heart symptoms
AlP
poisoning begins with severe metabolic acidosis and refractory hypotension.
These symptoms first cause shock and tissue perfusion failure due to
cardiogenic shock and peripheral circulatory failure two hours after
consumption, which marks the beginning of poisoning. Cardiovascular problems
such as acute myocardial infarctions and different cardiac arrhythmias are
significant and should be taken into consideration. According to post-mortem
accounts, hemodynamic instability, heart problems, and AlP poisoning causes
severe heart failure, non-specific localized necrosis, edema-induced separation
of myocardial fibers, eosinophil or
neutrophil infiltration, and vacuolation of myocytes (26).
ALP
poisoning frequently results in circulatory failure and severe hypotension, both
of which are major symptoms and causes of mortality. Due to the continued
absorption of PH3, hypotension, which is frequently severe and refractory, can
occur quickly and last for a long time. Arrhythmia, conduction issues,
myocardial injury, and myocardial depression can all contribute to intractable
shock. The extensive small vessel injury that causes peripheral circulatory
failure can also cause peripheral vasodilatation, which can result in shock.
Due to fluid loss, excessive vomiting may cause shock. Shock and a high
mortality rate can also result from the direct toxic effects of PH3 on the
adrenal cortex, which are accompanied by decreasing cortisol levels (27).
To
treat hypotension and refractory shock, medications like norepinephrine,
dopamine, phenylephrine, and dobutamine can be used. To manage cardiac
arrhythmias, anti-arrhythmic medications should be given. As an anti-ischemic
medication, trimetazidine has demonstrated outstanding results in stopping
ventricular ectopic beats and lowering oxygen consumption by converting
myocytes' metabolism from fatty acids to glucose. Recent research indicates
that the intra-aortic balloon pump (IABP) is an effective way to treat AlP
poisoning by mechanically maintaining the heart, particularly in cases of
refractory shock brought on by toxic myocarditis. The latest data support the
idea that digoxin administration can be employed to stabilize left ventricular
heart problems in AlP-poisoned cardiogenic shock by increasing myocardial
contractility and blood pressure (28).
Nervous
system
Headache,
fatigue, vertigo, weakness, paraesthesias, and drowsiness were some of the
neurological complaints. Long-term victims of PH3 poisoning may experience
severe headaches that don't go away and even peripheral neuropathy. Neuronal
degeneration, the removal of processes and Nissl granules, an eccentric
position of the degenerated nucleus, and the nucleolus are all examples of
neuropathological abnormalities. The production of PH3 gas, which interferes
with cellular oxygen use and causes neurocellular damage, may be the cause of
these hypoxic alterations (29).
The
primary target of ALP is the mitochondrial complex
More
than 90% of the total ATP needed by eukaryotic cells is supplied by
mitochondria. Phosphine interacts with the mitochondrial respiratory chain,
which is the primary source of free radical formation, by altering the electron
transfer chain. This interaction prevents oxidative phosphorylation, which
results in the high production of ROS and reduced ATP levels. A cell energy
crisis results from this. As a result, it is well recognized that mitochondria
are phosphine's primary target (30).
Cellular
oxidative stress functions similarly to reactive nitrogen species (RNS), which
are mostly comprised of NO and peroxynitrite as by-products of a group of
enzymes involved in electron transfer, in that it produces ROS such as
superoxide (O20) and H2O2. Cell death may result from ROS/RNS damaging biological
macromolecules. After entering the system, phosphorus interferes with the
creation of enzymes and proteins at the mitochondrial level. Additionally, the
generation of extremely reactive hydroxyl radicals plays a role in its
toxicology. The principal cause of ROS formation in the mitochondrial
respiratory chain is the reaction between the extremely reactive radical
phosphonate and this chain, which enters the intracellular space and disturbs
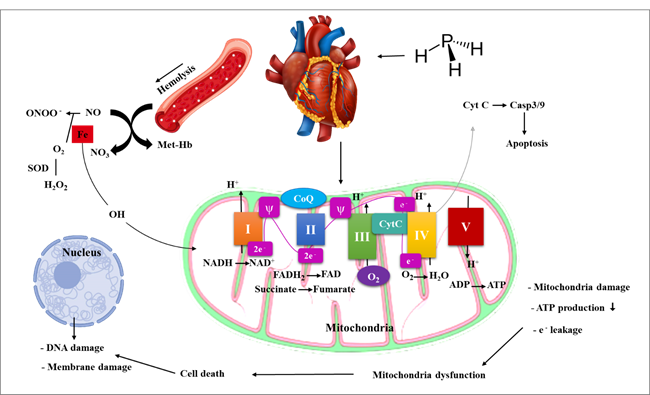
mitochondrial function (31) (Figure 1).
Figure 1. Phosphine functions by interfering
with the mitochondrial respiratory chain at the level of the mitochondria.
The
major sites of contact between phosphine and the electron transport chain are
Complex IV and cytochrome C oxidase. By suppressing this enzyme at the site of
Complex IV, phosphine decreases the chances of the mitochondrial membrane. In
addition, phosphine decreases the activity of complexes I and II, which in turn
decreases the activity of mitochondrial complexes and inhibits aerobic
respiration, causing a significant increase in ROS generation, reduced ATP
synthesis, and a loss of energy. A reduction in Ros generation and a reduction
in energy metabolism can enhance resistance to phosphine, while ROS generation
caused by phosphine poisoning is a deadly cause of energy deficit (18).
As a
result of phosphine's suppression of cytochrome oxidase, catalase and
peroxidase activity are reduced, hydrogen peroxide (H2O2) builds up, and
hydroxyl radicals (OH) are produced. While this is happening, ROS can harm or
change mitochondrial DNA, impairing respiration and overriding genes that
protect against phosphine poisoning. Increased phosphine resistance results
from the suppression of mitochondrial respiration chain genes, and its
persistence may be brought on by the activation of the genes that code for the
respiratory chain's constituents, Complexes I (NADH/ubiquinone) and III
(cytochrome c reductase). Gene complex III has a more significant function in
phosphine resistance as evidenced by the fact that resistance is increased when
this complex is reduced (32).
Phosphine
efficiency is linked to enhanced lipid peroxidation (LPO) after glutathione
(GSH) decreases in addition to boosting H2O2 generation. Since GSH catalyzes
H2O2 to O2 and H2O, which is a strategy to guard against oxidation, a decreased
concentration of GSH in many tissues in ALP poisoning can also explain cellular
damage (33). The elevation in free radicals
created by blocking respiratory chain complexes has been linked to cardiac
damage. These free radicals target the apoptotic process in cardiomyocytes by
causing LPO, DNA damage, and eventually oxidative stress . Furthermore, in the
event of PH3 poisoning, measuring cardiac damage markers like troponin can help
determine how quickly the heart is being damaged (34).
There
is proof that PH3 inhibits cytochrome c oxidase in vitro (complex IV). Since
PH3 inhibits cytochrome c oxidase activity less severely in vivo than in vitro,
this inhibition does not appear to be the main cause of toxicity. In contrast
to other cytochrome c oxidase inhibitors like cyanide, which significantly reduces
cytochrome c oxidase activity in vivo, PH3 does not do so. A decrease in
cytochrome c oxidase activity has also been observed in cyanide poisoning,
hemorrhagic shock, and sepsis (35). Hence, inhibition of cytochrome c
oxidase might not be the primary mechanism of its toxicity. Research on the
spectrum and dichroism have shown a connection to the heme moiety of cytochrome
oxidase. Additionally, it has been noted that PH3 interacts with hemoglobin's
heme to generate Heinz bodies. It is unknown if PH3 interacts with iron from
FeS centers, Cu cytochromes, and metal centers of enzymes in addition to iron
from heme. Studies that have also demonstrated a decline in the activity of
complexes I and II in rats lend weight to the idea that PH3 may affect the
function of other cytochromes and metalloproteins. Therefore, inhibiting the
electron transport chain (ETC) could lead to an increase in the formation of
reactive oxygen species (ROS) (36).
Applicability
of ALP and Intravascular Hemolysis
Organ
damage results from vascular wall degradation, hemolysis, and methemoglobinemia
(Met-Hb), which follow the intake of phosphine through the stomach mucosa. ALP
can damage blood vessels and the RBC membrane, or it can cause hemoglobinemia
and intravascular hemolysis via producing free radicals, where oxidative stress
significantly contributes to the development of these lesions (37).
Met-Hb
can be produced as a result of interaction with substances that oxidize ferrous
hemoglobin to ferric form. Multiple organ failure after exposure to ALP may
also be caused by decreased Met-ability Hb's to adequately oxygenate tissues.
According to recent studies, there is a strong and significant correlation
between blood levels of Met-Hb and death in poisoned persons. These
manifestations depend on blood concentration. The delivery of oxygen to the
target tissues may be significantly impacted by intravascular hemolysis.
Patients with Glucose-6-phosphate dehydrogenase (G6PD) deficiency frequently
have intravascular hemolysis, and when G6PD levels are low, erythrocytes'
capacity to generate NADPH is decreased and cells are more vulnerable to
hemolysis (38).
Metabolic
acidosis, a typical symptom of ALP poisoning, may also contribute to hemolysis.
Despite the frequent G6PD insufficiency that results from this poisoning, which
is caused by cardiogenic shock and mortality before hemolysis, hemolysis is
infrequently documented. Reduced G6PD also affects NO synthesis and heightens
vascular oxidative stress, which promotes the course of cardiovascular disease
(CVD) (39).
It
was discovered that PH3 inhibited insect catalase in three kinds of beetles
that were kept. Another study on insects found that superoxide dismutase (SOD),
a metalloenzyme, increased in activity in response to PH3 treatment whereas
catalase and peroxidase activity decreased. Superoxide anion (O2•) is
dismutated into hydrogen peroxide (H2O2), which is more stable and invasive, as
SOD activity is increased by PH3. The ability of catalase and peroxidase to
scavenge peroxide radicals is inhibited. The highly reactive hydroxyl radical
can then be created by hydrogen peroxide. This is in line with an elevation in
hydroxyl radical-related damage that has been seen in vitro, like lipid
peroxidation. The activity of antioxidant enzymes in humans may be impacted by
PH3, but this is unknown, as is whether the rise in SOD levels is controlled at
the transcriptional level (40, 41).
ALP's
impact on cardiomyocytes
The
major organ that is harmed by ALP poisoning is cardiac. During 12 to 24 hours
of exposure, cardiovascular problems brought on by ALP poisoning, such as
refractory hypotension, dysrhythmia, and heart problems, manifest. The main
causes of death in ALP poisoning have been recognized as cardiac toxicity,
cardiac dysfunction, and circulatory collapse that result in cardiomyocyte
death. Seventy-five percent of the heart's tissue is made up of cardiomyocytes,
which are crucial to the heart's blood flow. Cardiomyocytes include many
mitochondria, which are essential organelles. By making ATP through the process
of oxidative phosphorylation, mitochondria help cardiomyocytes contract and
generate 90% of their energy (42).
Impaired
hemostasis of cardiac energy is one of the most significant and obvious
symptoms of ALP poisoning, with ALP primarily damaging cardiac myocytes. ALP
also interferes with the electron transport chain, which interferes with cell
energy demand, inhibits the function of cytochrome c oxidase, an enzyme in the
ETC, lowers ATP levels, and eventually lowers myocardial energy. ALP-induced
cardiac toxicity is caused by lowering energy as well as the generation of free
radicals, particularly ROS, and oxidative stress, which results in LPO. Because
of its high oxygen intake, little amount of antioxidant system, and high
metabolic activity, the heart is generally particularly sensitive to oxidative
damage. Additionally, superoxide radical production and enzyme inhibition
decrease NO bioavailability (43, 44).
In
rare instances, alterations in biochemical indicators such as creatine
phosphokinase (CPK), creatine kinase myocardial band (CK-MB), and troponin-T
are also linked to ALP-induced myocardial injury. Some publications claim that
these biomarkers alter after ALP poisoning, while research conducted by
Soltaninejad and her team claims that these markers are inaccurate.
Additionally, despite ECG changes in acute poisoning, there are conflicting
reports of normal and abnormal levels of CPKMB. Despite the average level of
these enzymes, myocardial damage could still exist. As a result, it can be
concluded that in the future, with more research establishing the value of CPK
and CK-MB indicators as diagnostic markers, it will be feasible to forecast ALP
damage to cardiomyocytes and to stop the progression of the disease utilizing
the most effective treatment options (14).
The
majority of electrocardiographic (ECG) changes are nonspecific, including ST
and T-wave alterations that are likely caused by localized myocardial necrosis
and modifications to a membrane action potential. 80 percent of patients with
hypokinesia of the left ventricle and septum, 3 percent with akinesia, and
significantly lower ejection fractions were found in echocardiographic
investigations. Heart congestion, myocardial fiber separation and
fragmentation, nonspecific myocyte vacuolation, localized necrosis, and
neutrophil and eosinophil infiltration are common autopsy results. It makes
sense that the myocardial damage caused by PH3 is the cause of cardiac
dysfunction. Arrhythmias and abnormal conduction routes could occur as a result
of focal myocardial damage, whereas contractile dysfunction and hypotension
could ensue from broad myocardial damage (45).
The
exposure's biochemistry
Numerous
research has sought to concentrate on the specific mechanisms of metal
phosphide toxicity, notably on the direct impacts of PH3 gas, due to the
accessibility, common utilization, and terrible health consequences of
exposure. These research results can be divided into two areas that are loosely
correlated to one another, mitochondrial dysfunction and oxidative stress (36).
The
oxidative stress function
lipid
peroxidation and ROS—
Regarding
the impact of metal phosphides and their off-gas product PH3 on oxidative
stress, basic toxicological abnormalities have been documented. The effects of
phosphine gas have been linked to the production of reactive oxygen species
(ROS) and the suppression of detoxifying enzyme systems, according to chemical
models. Studies conducted in vitro have demonstrated that PH3 can convert Fe3+
to Fe2+ in cytochrome oxidase and cytochrome c. In the presence of hydrogen
peroxide (H2O2), 30 Fe2+ produces the ROS hydroxyl free radical (•OH), which is
essential for the creation of the highly unstable superoxide anions, •O2, as
well as a significant reactant and initiator of lipid peroxidation in Fenton
processes. Decreased glutathione (GSH) levels were shown to drop as a result of
phosphonate-induced oxidative damage in rats, while lipid peroxidation levels
in the liver, lungs, and brain rose. Studies on rats exposed to AlP provide
evidence for the presence of lipid peroxidation products in the brain. The rat
cerebellum, brain stem, and cerebrum all had decreased levels of total and
nonprotein sulfhydryls, according to Dua and Gill. Malondialdehyde (MDA), a
measure of lipid peroxidation, has increased significantly in rat cardiac
tissue after intragastric delivery of AlP. Additionally, after being exposed to
PH3 intraperitoneally, mouse liver MDA elevated. In a different investigation,
intraperitoneal injection of PH3 gas resulted in considerably increased MDA
concentrations in the rat brain, liver, and lung (36).
Rats
exposed to metal phosphide had a reduction in GSH as well as other aspects of
the GSH redox cycle, such as GSH reductase activity. Nevertheless, neither
catalase nor glutathione peroxidase changed in a different rat investigation
conducted by these same teams. Following exposure to AlP, patients' serum
enzyme activities of catalase and superoxide dismutase (SOD), which detoxify O2
to create H2O2, were both lowered and increased (46).
8-hydroxydeoxyguanosine
(8-OH-dGuo), an oxidation byproduct of DNA guanine, was raised by around 70% in
the brain and by 39% in the liver, which is an intriguing impact of PH3
poisoning. Increased levels of 8-OH-dGuo indicate that metal phosphide toxicity
affects nuclear and mitochondrial DNA (mtDNA) in addition to the simple target
organ, tissue, or cellular damage since it is a sensitive measure of ROS and a
key mutagen in DNA replication. Following their metabolic conversion to
reactive nucleophiles, electrophilic substances might develop inherent
mutagenesis properties. The long-term implications on the downstream gene
expression in the instance of metal phosphide-induced mutagenesis have not been
experimentally investigated or epidemiologically (47).
Mitochondria
function
The
functions of mitochondria in producing energy, controlling redox, maintaining
calcium homeostasis, and intermediate metabolism are well established. Exposure
to metal phosphide can have extremely hazardous effects, and some of those
symptoms may be brought on by impaired metabolic processes. For instance,
numerous studies have demonstrated that the disruption or inhibition of
mitochondrial function, namely the suppression of cytochrome c oxidase
activity, may result in toxicity (48).
In
contrast to their function in energy metabolism, mitochondria also contribute
significantly to the generation of ROS and the activation of cell death-related
pathways. The intrinsic pathway of programmed cell death, as opposed to the
extrinsic pathway, which is primarily driven by extracellular inputs, involves
mitochondrial signaling. Numerous studies that have been published recently
suggest that ROS are crucial to pathophysiological processes, particularly when
considering the function of mitochondrial cell signaling and biological
consequence. The majority of intrinsic detoxification enzymatic activities,
including manganese SOD, remove or render harmless ROS, which are by-products
of cellular respiratory activity (MnSOD) (49).
In
reality, undamaged mitochondria are necessary for the matrix enzyme MnSOD to
convert O2 to H2O2. In MnSOD mutant mice, loss of control over O2 and H2O2
generation has been demonstrated to be lethal. Increased ROS generation has the
potential to swiftly overwhelm the detoxifying mechanism if active electron
transport through the respiratory chain is interrupted. In comparison to the
cytosol and nuclear regions, steady-state oxygen levels in the mitochondria can
be up to 10-fold greater, according to Cadenas and Davies. This is a
significant source of ROS that is ready to be released in the event of an
improper signal (50).
In
addition to being a significant generator of H2O2 and •O2, mitochondria also
act as targets. By altering mitochondrial proteins, lipids, and DNA, the
resulting oxidative damage can cause bioenergetic abnormalities and the start
of cell death. 43 Complexes I, II, and III-embedded Rieske Fe-S clusters, which
are oxidatively damaged areas of the respiratory chain, can sometimes enhance
the generation of oxygen by a factor of four. MnSOD quickly converts
mitochondrial matrix O2 into H2O2, which can diffuse past membranes and into
the cytoplasm, where it can function as a second messenger in the control of
NF-B. The proinflammatory cascade, which consists of TNF, MIP-2, and IL-8, has
been demonstrated to depend on NF-B. In humans, significant metabolic diseases
that ultimately endanger survival can be caused by flaws in oxidation,
phosphorylation, and/or anomalies along any part of the aforementioned
respiratory chain complex. An excellent and thorough review of mitochondria and
ROS production (51).
Complex
I is thought to be the main source of electron leaks, resulting in the release
of O2.47 Increased ROS and/or intracellular Ca2+ concentrations can negatively
affect the regulation of MPTP if their neutralizing systems are unable to keep
up with them. Apoptotic proteins are enhanced as a result of Δψm opening, and
cytochrome c triggers more caspases into the cellular environment. In cultured
hepatocytes, trichlorfon led to the release of cytochrome c from the
mitochondria, activating caspase 3. When oxidative damage to cells or tissues
occurs, harmful chemicals other than caspases are also released. The cytokine TNF
can promote either cell death or life by acting on particular receptors. An
increase in mitochondrial ROS activity in a human embryonic kidney cell line
after TNF treatment suggests that impaired mitochondrial metabolism may result
from the extramitochondrial production of inflammatory cytokines. The buildup
of electrons from highly reactive carriers and the loading of the cell with
extra ROS can both be caused by inhibition along specific segments of the
respiratory chain. Cardiolipin, a mitochondrial lipid produced in response to
TNF-induced caspase 8 activation, can be oxidized by additional ROS (52).
To
maintain certain cytosolic Ca2+ gradients, mitochondria typically build up and
release Ca2+ over time. This process is essential for cell survival. However,
highly high intramitochondrial Ca2+ can overwhelm the remaining undamaged
mitochondria when discharged into the cytosol as a result of damage, leading to
the collapse of the electrochemical gradient across the inner membrane and the
inability to make ATP. It is believed that endothelial cells' ryanodine
receptors on the endoplasmic reticulum cause Ca2+ release in response to
mitochondria-derived ROS (52).
Numerous
studies demonstrate that metal phosphides/PH3 attack affects mitochondrial
metabolic balance and detoxifying mechanisms. In isolated mouse liver
mitochondria, PH3 has been demonstrated to inhibit complex IV (cytochrome c
oxidase) of the respiratory chain and reduce the potency of Δψm. There is
evidence that metal phosphide exposure affects the brain and liver's balance of
glucose. High metabolic rates are needed by the brain, which also depends on
glucose to maintain neuronal activity. The rats exposed to acute levels of AlP
have enhanced lactate dehydrogenase (LDH) activity in the brain (53).
This
strongly suggests that aerobic metabolism has changed to anaerobic metabolism
and denotes serious mitochondrial malfunction. Elevation in LDH activity,
according to research, leads to the reversible conversion of pyruvate to
lactate and the reoxidation of NADH (nicotinamide adenine dinucleotide reduced
form) to NAD+ (NAD oxidized form), both of which are independent of
mitochondrial electron transport. When this happens, lactate builds up in the
brain, which puts animals at risk for problems with CNS-related activities.
Both rodents' and insects' liver mitochondria activity is significantly
influenced by PH3. Both create H2O2 after being challenged with PH3. Insect
mitochondrial myxothiazol and antimycin inhibition of respiratory chain
activity revealed that glycerophosphate dehydrogenase auto-oxidation is the
source of H2O2 generation after PH3 exposure. PH3 inhibits the cytochrome c
cascade's electron transport, but cyanide—another well-known metabolic toxin
and a well-known example of a respiratory chain toxicant—blocks the passage of
electrons from cytochrome a and a3 to oxygen. However, further research
revealed that, in some circumstances, PH3 predominantly inhibits one component
of the cytochrome aa3 complex (complex IV), such as cytochrome a. This
inhibition may be caused by PH3 altering the valence state of heme iron (54).
The
availability and metabolic absorption of oxygen have been directly connected to
the toxicity of PH3. In the presence of oxygen, toxicity rises, whereas, in anoxic
conditions, it falls. In contrast to anaerobic settings, the toxicity of PH3
rises in more aerobic environments (36) (Figure 2).
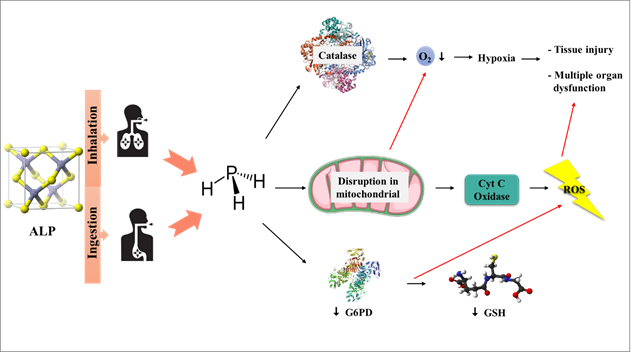
Figure 2. Aluminum phosphide releases
phosphine gas when exposed to moisture and/or stomach acid due to oxidative
damage.
Conclusions
Metal phosphide poisoning,
particularly aluminum phosphide (ALP), is one of the major health hazards
facing modern human societies. Numerous studies have been conducted on the
pathophysiology of ALP in patients, and the majority of these studies' findings
highlight the fact that ALP leads to mitochondrial dysfunction.RBC
intravascular hemolysis and ATP generation are both diminished by mitochondrial
failure. It has been established that irreparably damaged metabolism is a
component of PH3 poisoning. Mechanistically, this might happen due to a
metabolic crisis or as an indirect result of increased ROS production brought
on by the metabolism, which ultimately causes cellular/target organ collapse.
Overall, this has had a seriously negative impact on the survival rates of
those who were exposed. Myocyte performance is lowered as a consequence of
these diseases, and CVD results. It might be argued that finding indicators
linked to cardiac diseases in the early stages of sickness would be beneficial
because cardiac ailments, particularly cardiac shock, are the major causes of
death in ALP patients. A more logical approach to treatment and the
identification of temporal therapeutic windows will be made possible with
further research into the precise correlations between exposure, metabolic, and
systemic toxicity.
Author contribution
MRT and HMK designed the project, wrote the manuscript and analyzed the data.
All the authors read and confirmed the final edited version of the manuscript.
Conflict of interest
The authors declare that they have no conflicts of interest.
References
1. Gurjar
M, Baronia AK, Azim A, Sharma K. Managing aluminum phosphide poisonings. J
Emerg Trauma Shock. 2011;4(3):378-84.
2. Sedaghattalab M. Treatment of
critical aluminum phosphide (rice tablet) poisoning with high-dose insulin: a
case report. J Med Case Rep. 2022;16(1):192.
3. Singh S, Bhalla A, Verma SK,
Kaur A, Gill K. Cytochrome-c oxidase inhibition in 26 aluminum phosphide
poisoned patients. Clin Toxicol (Phila). 2006;44(2):155-8.
4. Goel A, Aggarwal P. Pesticide
poisoning. Natl Med J India. 2007;20(4):182-91.
5. Tiwary AK, Puschner B,
Charlton BR, Filigenzi MS. Diagnosis of zinc phosphide poisoning in chickens
using a new analytical approach. Avian Dis. 2005;49(2):288-91.
6. Mohan B, Gupta V, Ralhan S,
Gupta D, Puri S, Wander GS, et al. Role of Extracorporeal Membrane Oxygenation
in Aluminum Phosphide Poisoning-Induced Reversible Myocardial Dysfunction: A
Novel Therapeutic Modality. J Emerg Med. 2015;49(5):651-6.
7. Dadpour B, Mokhtarpour M,
Abdollahi M, Afshari R. An outbreak of aluminium phosphide poisoning in
Mashhad, Iran. Arh Hig Rada Toksikol. 2016;67(1):65-6.
8. Salimi A, Jamali Z, Shabani M.
Antioxidant Potential and Inhibition of Mitochondrial Permeability Transition
Pore by Myricetin Reduces Aluminium Phosphide-Induced Cytotoxicity and
Mitochondrial Impairments. Front Pharmacol. 2021;12:719081.
9. Farahani MV, Soroosh D,
Marashi SM. Thoughts on the current management of acute aluminum phosphide
toxicity and proposals for therapy: An Evidence-based review. Indian J Crit
Care Med. 2016;20(12):724-30.
10. Moghadamnia AA. An update on
toxicology of aluminum phosphide. Daru. 2012;20(1):25.
11. Pannu AK. Pulmonary Management
in Aluminum Phosphide Poisoning. Indian J Crit Care Med. 2017;21(1):63-4.
12. Bagheri-Moghaddam A, Abbaspour
H, Tajoddini S, Mohammadzadeh V, Moinipour A, Dadpour B. Using Intra-Aortic
Balloon Pump for Management of Cardiogenic Shock Following Aluminum Phosphide
Poisoning; Report of 3 Cases. Emerg (Tehran). 2018;6(1):e3.
13. Gadour E, Kotb A. Systematic
Review of Antifungal-Induced Acute Liver Failure. Cureus. 2021;13(10):e18940.
14. Soltaninejad K, Beyranvand MR,
Momenzadeh SA, Shadnia S. Electrocardiographic findings and cardiac
manifestations in acute aluminum phosphide poisoning. J Forensic Leg Med.
2012;19(5):291-3.
15. Kim BS, Li BT, Engel A, Samra
JS, Clarke S, Norton ID, et al. Diagnosis of gastrointestinal bleeding: A
practical guide for clinicians. World J Gastrointest Pathophysiol.
2014;5(4):467-78.
16. Dorooshi G, Mirzae M, Fard NT,
Zoofaghari S, Mood NE. Investigating the Outcomes of Aluminum Phosphide
Poisoning in Khorshid Referral Hospital, Isfahan, Iran: A Retrospective Study.
J Res Pharm Pract. 2021;10(4):166-73.
17. Dhondup T, Qian Q. Electrolyte
and Acid-Base Disorders in Chronic Kidney Disease and End-Stage Kidney Failure.
Blood Purif. 2017;43(1-3):179-88.
18. Anand R, Kumari P, Kaushal A,
Bal A, Wani WY, Sunkaria A, et al. Effect of acute aluminum phosphide exposure
on rats: a biochemical and histological correlation. Toxicol Lett.
2012;215(1):62-9.
19. Hugar BS, Praveen S, Hosahally
JS, Kainoor S, Shetty AR. Gastrointestinal hemorrhage in aluminum phosphide
poisoning. J Forensic Sci. 2015;60 Suppl 1:S261-3.
20. Saleki S, Ardalan FA,
Javidan-Nejad A. Liver histopathology of fatal phosphine poisoning. Forensic
Sci Int. 2007;166(2-3):190-3.
21. Jafari A, Baghaei A, Solgi R,
Baeeri M, Chamanara M, Hassani S, et al. An electrocardiographic, molecular and
biochemical approach to explore the cardioprotective effect of vasopressin and
milrinone against phosphide toxicity in rats. Food Chem Toxicol.
2015;80:182-92.
22. Tehrani H, Halvaie Z, Shadnia
S, Soltaninejad K, Abdollahi M. Protective effects of N-acetylcysteine on
aluminum phosphide-induced oxidative stress in acute human poisoning. Clin
Toxicol (Phila). 2013;51(1):23-8.
23. Agrawal VK, Bansal A, Singh RK,
Kumawat BL, Mahajan P. Aluminum phosphide poisoning: Possible role of
supportive measures in the absence of specific antidote. Indian J Crit Care
Med. 2015;19(2):109-12.
24. Mirakbari SM. Proposal for a
new mechanism of action for aluminum phosphide (ALP) for causing local injuries
in ALP poisoning: Should treatment strategies be modified? Hum Exp Toxicol.
2016;35(10):1145-6.
25. Mehrpour O, Jafarzadeh M,
Abdollahi M. A systematic review of aluminium phosphide poisoning. Arh Hig Rada
Toksikol. 2012;63(1):61-73.
26. Oghabian Z, Mehrpour O.
Treatment of Aluminium Phosphide Poisoning with a Combination of Intravenous
Glucagon, Digoxin and Antioxidant Agents. Sultan Qaboos Univ Med J.
2016;16(3):e352-5.
27. Sahoo D, Kujur ST, Das DS, Dey
A, Devi S. Aluminium Phosphide Poisoning: Early Suspicion of Cardiotoxicity Is
Necessary for Improved Outcomes. Cureus. 2020;12(9):e10237.
28. Siddaiah L, Adhyapak S, Jaydev
S, Shetty G, Varghese K, Patil C, et al. Intra-aortic balloon pump in toxic
myocarditis due to aluminum phosphide poisoning. J Med Toxicol. 2009;5(2):80-3.
29. Shafiee Dolat Abadi S, Zamani
N, Abbasi S, Shojaei M, Hassanian-Moghaddam H. Neurologic sequelae of phosphide
poisoning: A case report. Front Neurol. 2022;13:888493.
30. Baghaei A, Solgi R, Jafari A,
Abdolghaffari AH, Golaghaei A, Asghari MH, et al. Molecular and biochemical
evidence on the protection of cardiomyocytes from phosphine-induced oxidative stress,
mitochondrial dysfunction and apoptosis by acetyl-L-carnitine. Environ Toxicol
Pharmacol. 2016;42:30-7.
31. Kariman H, Heydari K, Fakhri M,
Shahrami A, Dolatabadi AA, Mohammadi HA, et al. Aluminium phosphide poisoning
and oxidative stress: serum biomarker assessment. J Med Toxicol.
2012;8(3):281-4.
32. Nath NS, Bhattacharya I, Tuck
AG, Schlipalius DI, Ebert PR. Mechanisms of phosphine toxicity. J Toxicol.
2011;2011:494168.
33. Jagadeesan R, Collins PJ,
Daglish GJ, Ebert PR, Schlipalius DI. Phosphine resistance in the rust red
flour beetle, Tribolium castaneum (Coleoptera: Tenebrionidae): inheritance,
gene interactions and fitness costs. PLoS One. 2012;7(2):e31582.
34. Haghi Aminjan H, Abtahi SR,
Hazrati E, Chamanara M, Jalili M, Paknejad B. Targeting of oxidative stress and
inflammation through ROS/NF-kappaB pathway in phosphine-induced hepatotoxicity
mitigation. Life Sci. 2019;232:116607.
35. Bogale DE, Ejigu BD, Muche TA.
Clinical Profile and Treatment Outcome of Aluminum Phosphide Poisoning in
Felege Hiwot Referral Hospital, Northwest Ethiopia: A Retrospective Study. Open
Access Emerg Med. 2021;13:239-48.
36. Sciuto AM, Wong BJ, Martens ME,
Hoard-Fruchey H, Perkins MW. Phosphine toxicity: a story of disrupted
mitochondrial metabolism. Ann N Y Acad Sci. 2016;1374(1):41-51.
37. Vosooghi AA, Salmasi M. G6PD
deficiency and aluminum phosphide poisoning. J Res Med Sci. 2018;23:83.
38. Lehoux J, Hena Z, McCabe M,
Peek G. Aluminium phosphide poisoning resulting in cardiac arrest, successful
treatment with Extracorporeal Cardiopulmonary resuscitation (ECPR): a case
report. Perfusion. 2018;33(7):597-8.
39. Kuhn V, Diederich L, Keller
TCSt, Kramer CM, Lückstädt W, Panknin C, et al. Red Blood Cell Function and
Dysfunction: Redox Regulation, Nitric Oxide Metabolism, Anemia. Antioxid Redox
Signal. 2017;26(13):718-42.
40. Doukyu N, Taguchi K.
Involvement of catalase and superoxide dismutase in hydrophobic organic solvent
tolerance of Escherichia coli. AMB Express. 2021;11(1):97.
41. Bian Y, Rong Z, Chang TM.
Polyhemoglobin-superoxide dismutase-catalase-carbonic anhydrase: a novel
biotechnology-based blood substitute that transports both oxygen and carbon
dioxide and also acts as an antioxidant. Artif Cells Blood Substit Immobil
Biotechnol. 2012;40(1-2):28-37.
42. Asghari MH, Moloudizargari M,
Baeeri M, Baghaei A, Rahimifard M, Solgi R, et al. On the mechanisms of
melatonin in protection of aluminum phosphide cardiotoxicity. Arch Toxicol.
2017;91(9):3109-20.
43. Goharbari MH, Taghaddosinejad
F, Arefi M, Sharifzadeh M, Mojtahedzadeh M, Nikfar S, et al. Therapeutic
effects of oral liothyronine on aluminum phosphide poisoning as an adjuvant
therapy: A clinical trial. Hum Exp Toxicol. 2018;37(2):107-17.
44. Gouda AS, El-Nabarawy NA,
Ibrahim SF. Moringa oleifera extract (Lam) attenuates Aluminium
phosphide-induced acute cardiac toxicity in rats. Toxicol Rep. 2018;5:209-12.
45. Guru S, Kumar R, Behera A,
Patra S, Kumar P, Jr. Aluminium Phosphide-Induced Expression of Covertly
Present Brugada Pattern in Electrocardiogram: A Rare Case Report. Cureus.
2020;12(9):e10552.
46. Żebrowska E, Maciejczyk M,
Żendzian-Piotrowska M, Zalewska A, Chabowski A. High Protein Diet Induces Oxidative
Stress in Rat Cerebral Cortex and Hypothalamus. Int J Mol Sci. 2019;20(7).
47. Solgi R, Baghaei A, Golaghaei
A, Hasani S, Baeeri M, Navaei M, et al. Electrophysiological and molecular
mechanisms of protection by iron sucrose against phosphine-induced
cardiotoxicity: a time course study. Toxicol Mech Methods. 2015;25(4):249-57.
48. Jiang J, Liang S, Zhang J, Du
Z, Xu Q, Duan J, et al. Melatonin ameliorates PM(2.5) -induced cardiac
perivascular fibrosis through regulating mitochondrial redox homeostasis. J
Pineal Res. 2021;70(1):e12686.
49. Marchi S, Giorgi C, Suski JM,
Agnoletto C, Bononi A, Bonora M, et al. Mitochondria-ros crosstalk in the
control of cell death and aging. J Signal Transduct. 2012;2012:329635.
50. Dikalov S. Cross talk between
mitochondria and NADPH oxidases. Free Radic Biol Med. 2011;51(7):1289-301.
51. Chan CM, Huang DY, Sekar P, Hsu
SH, Lin WW. Reactive oxygen species-dependent mitochondrial dynamics and
autophagy confer protective effects in retinal pigment epithelial cells against
sodium iodate-induced cell death. J Biomed Sci. 2019;26(1):40.
52. Zorov DB, Juhaszova M, Sollott
SJ. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release.
Physiol Rev. 2014;94(3):909-50.
53. Dua R, Gill KD. Effect of
aluminium phosphide exposure on kinetic properties of cytochrome oxidase and
mitochondrial energy metabolism in rat brain. Biochim Biophys Acta.
2004;1674(1):4-11.
54. Yaku K, Okabe K, Gulshan M,
Takatsu K, Okamoto H, Nakagawa T. Metabolism and biochemical properties of nicotinamide
adenine dinucleotide (NAD) analogs, nicotinamide guanine dinucleotide (NGD) and
nicotinamide hypoxanthine dinucleotide (NHD). Sci Rep. 2019;9(1):13102.